钝药野木瓜中三萜类化合物分离及其含量测定
2018-03-29*
*
1.贵阳中医学院药学系,贵州 贵阳 550002;2.贵州师范大学分析测试中心, 贵州 贵阳 550001;3.黔南州瓮安县人民医院, 贵州 黔南 550400
钝药野木瓜(StauntonialeucanthaDiels ex Y.C.)属于木通科野木瓜属植物,俗称绕绕藤、五叶木通、八月瓜,主要分布在贵州、四川、安徽、江浙和两广一带,具有祛风止痛,利尿消肿的功效,对风湿痹通、小便不利和水肿疗效显著[1]。该药材被2003版《贵州省中药材、民族药材质量标准》收载,作为野木瓜藤入药,但仅有性状描述[2]。在长期研究抗病毒中药和民族药的过程中,采用HIV-1蛋白酶和HPLC法[3],发现钝药野木瓜95%乙醇提取物的乙酸乙酯萃取物在100 μg/mL时,对HIV蛋白酶的抑制率达44.3%,进一步的文献调研发现,钝药野木瓜化学成分及含量研究未见报道。2015 版《中国药典》同属植物野木瓜(S.chinensisDC)以苯乙醇苷类成分荷苞花苷B作为指标性成分进行质量控制[4]。本课题组参照药典方法对黔产10批次的钝药野木瓜药材中荷苞花苷B进行定性和定量研究,发现含量为5.91~39.39 μg/g,荷苞花苷B需低温存放,长时间暴露在空气中,含量有所下降,其标准品溶液低温存放超过24 h后,易产生杂质峰[5]。一些学者对野木瓜(S.chinensisDC)中的木犀草素、绿原酸、齐墩果酸和熊果酸的含量进行了报道[6-8],而同属植物化学成分研究发现野木瓜(S.chinensisDC)[9]、六叶野木瓜(S.hexaphyllaThunb.Dence.)、五指那藤(S.obovatifoliolasubsp.intermedia)、尾叶那藤(S.obovatifoliolasp.urophylla)和黄腊果(S.brachyanthera)藤茎中常分离出羽扇豆醇和羽扇豆酮等多种羽扇豆烷型化合物[9-11]。体内外研究证实,羽扇豆醇具抗炎、抗疟疾、抗癌等作用[12]。羽扇豆酮对单纯疱疹病毒(HSV)和非洲猪瘟病毒(ASFV)具有抗病毒活性,对HSV-1和HSV-2具有很强的抗病毒抑癍作用[13]。为明确钝药野木瓜药效物质基础,实验开展了钝药野木瓜化学成分研究,建立HPLC法同时测定羽扇豆醇和羽扇豆酮的定量分析方法,为该药材的监督、质控和相关药品、保健品的开发研制提供科学依据。
1 仪器与材料
1.1 仪器 Bruker AM-400和500 MHz核磁共振波谱仪(TMS为内标);GC-MS 5973型气相色谱质谱联用仪(美国HP公司);1100型高效液相色谱仪( 美国 Agilent);梅特勒-托利多超声波清洗器(METILER-TOULEDO);ME104 电子分析天平(瑞士梅特勒);R-200旋转蒸发仪(瑞士布奇公司);SHZ-DⅢ循环水式真空泵(上海予英仪器有限公司);ZF-1三用紫外分析仪(上海电光分析)。
1.2 材料 TLC和柱色谱硅胶分别为GF254和200-300目硅胶 (青岛海洋化工厂分厂);反相硅胶为ODS DM 1020T (日本富士硅胶公司);Sephadex LH-20 (GE 公司)。羽扇豆醇、羽扇豆烷均来自为本植物(乙酸乙酯萃取)分离纯化所得,结构经波谱学(ESI-MS,1H-NMR,13C-NMR)分析鉴定,以HPLC面积归一化法测定其质量分数均大于96%。洗脱剂均为工业重蒸品,甲醇、乙醇、磷酸(分析纯,重庆茂业化学试剂有限公司);乙腈(色谱纯,天津市科密欧化学试剂有限公司);水(超纯水)。钝药野木瓜药材于2014年4月采集于贵州省遵义市,经贵阳中医学院陈德媛教授鉴定为钝药野木瓜的地上部分,凭证标本保存于贵阳中医学院中药化学教研室(标本号:2014/SL)。
2 方法与结果
2.1 化学成分研究
2.1.1 药材前处理 干燥钝药野木瓜藤茎 3 kg,粉碎后用70%的甲醇溶液(料液比为 1∶3)加热回流提取3次,每次1 h,合并浸提液后减压浓缩,回收甲醇至无醇味,再用乙酸乙酯萃取,浓缩,得浸膏(45 g),经硅胶柱(柱长×内径=75 cm × 5.5 cm )色谱进行分离。以石油醚-乙酸乙酯(40∶1 → 0∶100)进行梯度洗脱,最后用氯仿-甲醇(1∶1)混合液及纯甲醇下柱,得 35 个流份(fr.1~ 35)。
2.1.2 化合物的分离纯化 在fr.3,即 石油醚-乙酸乙酯 25∶1 部分(112 mg)经Sephadex LH-20凝胶柱色谱分离,以氯仿-甲醇(1∶1)为洗脱剂,纯化得到化合物 1(27 mg);fr. 18,即石油醚-乙酸乙酯 20∶1~18∶1部分(106 mg)经反相硅胶柱层析,以甲醇-水(5∶5 → 10∶0)梯度洗脱,在 8∶2 洗脱部分析出结晶,用甲醇多次洗涤结晶,石油醚-丙酮重结晶得化合物2(19 mg);fr.26,即石油醚-乙酸乙酯(15∶1 ~ 12∶1部分(363 mg)经硅胶柱层析分离,用石油醚-丙酮(30∶1→1∶1)梯度洗脱得到 12 个流份,其中fr.5 ~ 6经Sephadex LH-20柱层析,甲醇洗脱分离纯化得到化合物 3(26.7 mg);fr.30,即石油醚-乙酸乙酯10∶1~9∶1部分(163 mg)经硅胶柱层析,用氯仿-丙酮(40∶1→1∶1)洗脱,并经石油醚-丙酮重结晶得到化合物 4(25.2 mg)
2.1.3 化合物的结构鉴定 化合物 1 白色结晶(丙酮),mp 170~172 ℃,分子式为C30H48O; ESI-MSm/z447 [M + 23]+;1H-NMR (CDCl3, 400 MHz) 、13C-NMR (CDCl3, 100 MHz) 数据与文献[14]对照基本一致,故鉴定化合物 1 为羽扇豆酮。
化合物 2 白色针晶(石油醚-丙酮);EI-MSm/z488 [M]+,1H-NMR (CDCl3,500 MHz)、13C-NMR (CDCl3, 125 MHz)数据与文献[15]报道基本一致,故鉴定该化合物2为3β-乙酰基齐墩果酸。
化合物 3 白色针状结晶(石油醚-醋酸乙酯), mp 210 ~ 212 ℃,分子式为C30H50O, TLC检识遇硫酸-甲醇溶液加热显紫红色, Lieberman-Burchard反应为阳性。EI-MSm/z426 [M]+,1H-NMR (CDCl3, 400 MHz) 、13C-NMR (CDCl3, 100 MHz)数据与文献[16]对照基本一致,故鉴定化合物 3 为羽扇豆醇。
化合物 4 白色簇晶(石油醚-丙酮);EI-MSm/z456 [M]+,1H-NMR (CDCl3, 400 MHz) 、13C-NMR (CDCl3, 100 MHz)数据与文献[17]报道基本一致,故鉴定该化合物4为齐墩果酸。
2.2 羽扇豆醇与羽扇豆酮的含量测定
2.2.1 色谱条件及系统适应性 色谱柱为Ocean C18柱(250 mm × 4.6 mm,5 μm),流动相:100%甲醇;流速:1 mL/min;柱温:30 ℃;紫外检测波长:210 nm;进样量为20 μL。对照品与样品中的羽扇豆醇和羽扇豆酮色谱峰相互分离度大于1.5,理论塔板数大于5000,对照品和钝药野木瓜样品的HPLC色谱见图 1。

2.2.2 对照品溶液的制备 精密称取羽扇豆醇对照品12.930 mg和羽扇豆酮对照品23.625 mg于25 mL容量瓶中,加甲醇溶液溶解并稀释至刻度,制成浓度分别为0.5172 mg/mL和0.945 mg/mL对照品贮备液。分别精密吸取上述对照品贮备液适量,加甲醇溶解并稀释,摇匀,制成羽扇豆醇和羽扇豆酮质量浓度分别为0.0517 mg/mL和0.0945 mg/mL的混合对照品溶液,备用。
2.2.3 供试品溶液的制备 取钝药野木瓜干燥带叶茎枝药材粉末(贵州都匀,过4号筛,50℃烘干)约1.0 g,精密称定,置于150 mL具塞锥形瓶中,用移液管精密加入甲醇25 mL,密塞,摇匀,称重,超声提取30 min,待冷却后,称重,用甲醇补足减量,经4000 r/min离心15 min,取上清液过0.45 μm 微孔滤膜,即得。
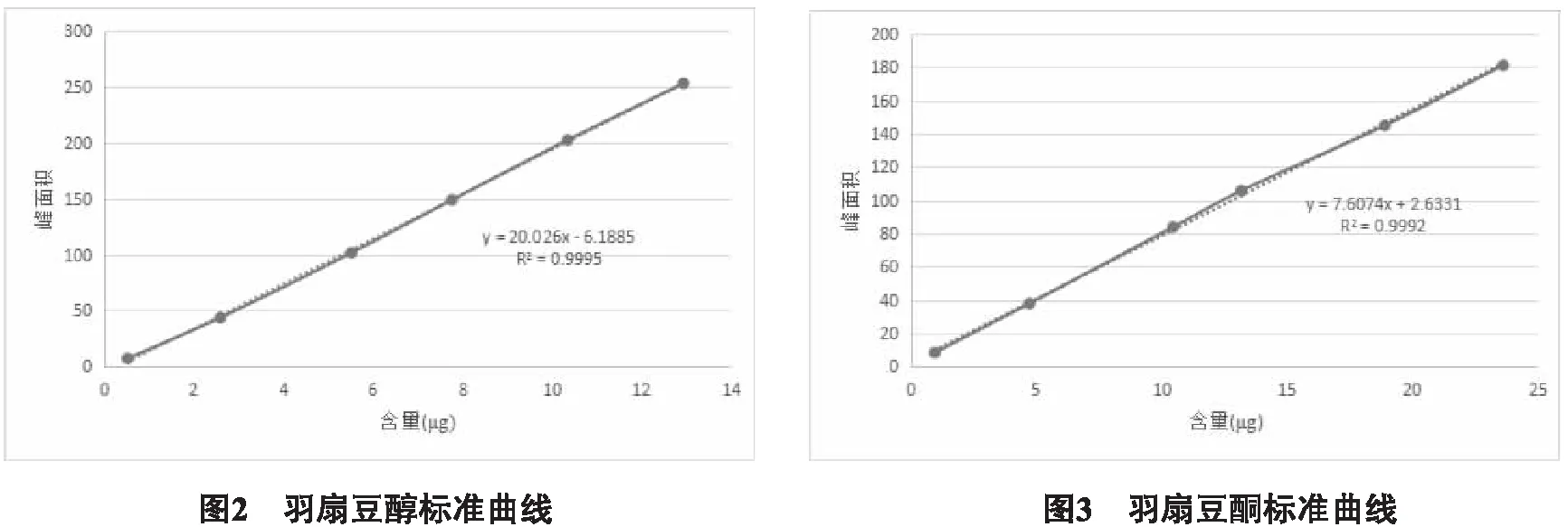
2.2.4 标准曲线的考察 分别精密吸取上述两种对照品贮备液1、5、10、15、20、25 μL,按2.2.1项下色谱条件分别进样,记录色谱图,以进样量(X,μg)为横坐标,峰面积积分值(Y)为纵坐标进行回归,得羽扇豆醇回归方程为Y=20.026X-6.1885(r=0.9995,n=6),进样量在0.5172~12.93 μg范围内线性关系良好;回归方程为羽扇豆酮Y=7.6074X+2.6331(r=0.9992,n=6),进样量在0.945~23.625 μg范围内线性关系良好。见图2~3。
2.2.5 精密度试验 分别精密吸取2.2.2项下对照品混合溶液20 μL,按2.2.1色谱条件各连续进样6次,测定峰面积,计算RSD分别为0.9%和0.3%,表明仪器的精密度良好。
2.2.6 重复性试验 精密称取同一批钝药野木瓜粉末(贵州都匀,过4号筛)6份,每份1.0 g,按供试品溶液的制备方法制备溶液,取供试品溶液20 μL进样,在上述色谱条件下进行分析,根据峰面积代入回归方程计算质量,得羽扇豆醇和羽扇豆酮RSD分别为1.2%(n=6)和0.8%(n=6)。
2.2.7 稳定性试验 精密称取钝药野木瓜粉末(贵州都匀,过4号筛)约1.0 g,按2.2.3项下方法制备供试品溶液,室温下放置,取供试品溶液20μL,分别在0、2、4、6、8、10、12 h进样分析,分别测定峰面积。结果羽扇豆醇和羽扇豆酮峰面积的RSD分别为2.0%和1.5%。表明12 h内供试品溶液的稳定性良好。
2.2.8 加样回收率试验 精密称取钝药野木瓜粉末(贵州都匀,过4号筛)约0.5 g,各加入对照品适量,按2.2.3项下方法制备样品溶液,分别吸取20 μL进样分析,测定羽扇豆醇、羽扇豆酮的含量,计算加样回收率。见表1。
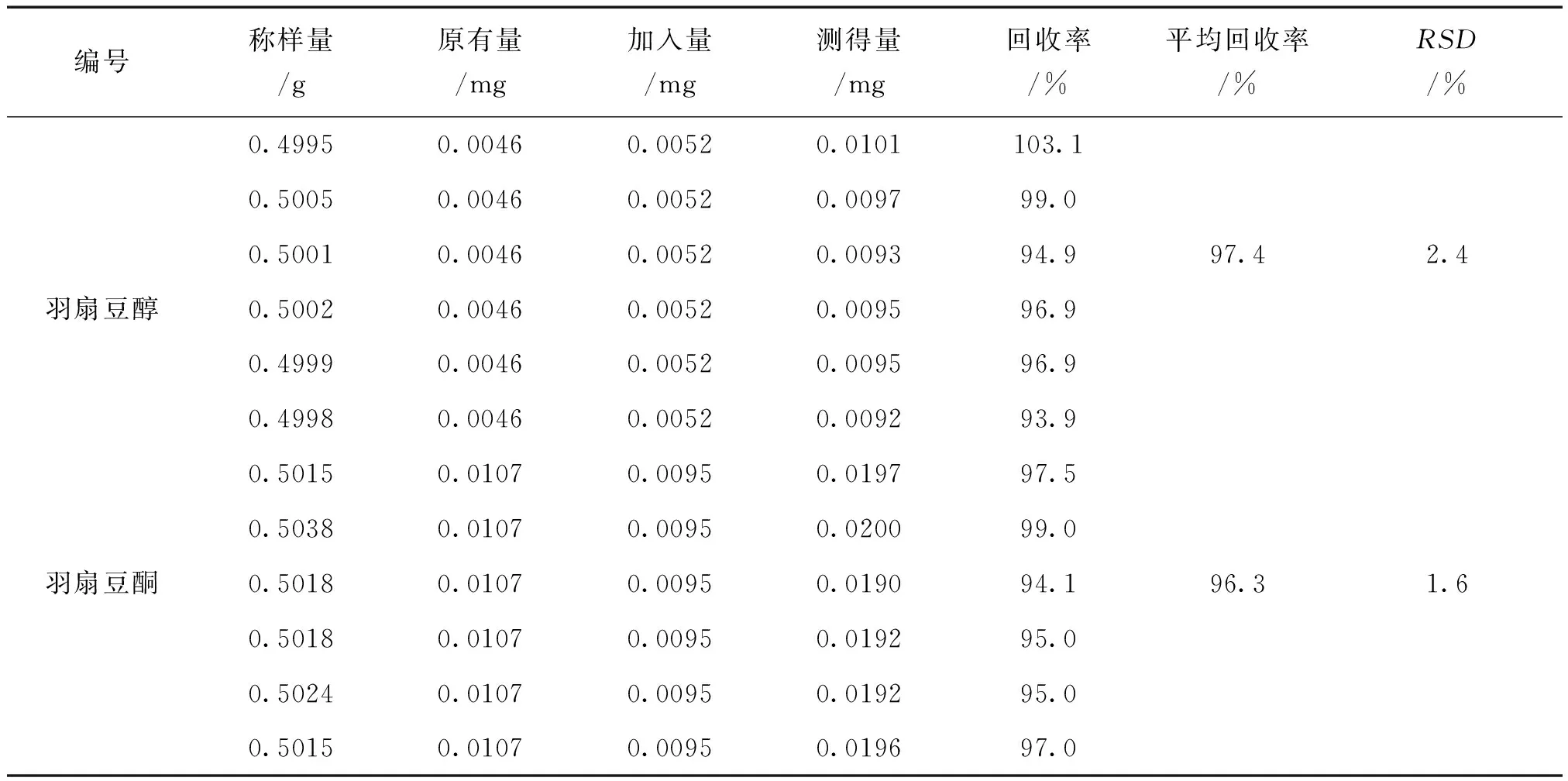
表1 钝药野木瓜加样回收率结果
2.2.9 样品分析 取贵州省3个地区钝药野木瓜的茎和全株,8个地区钝药野木瓜的全株,打成粉末(过4号筛)精密称定1.0 g,按2.2.3项下方法制备供试品溶液,按2.2.1项下色谱条件进样,记录峰面积,外标一点法计算药材中羽扇豆醇和羽扇豆酮的含量。实验结果见表2~3。

表2 钝药野木瓜药材不同采收部位两种三萜含量检测结果 (n=3)

表3 贵州8个地区钝药野木瓜药材中两种三萜含量检测结果 (n=10)
3 讨论
3.1 实验条件的优化 实验考察了甲醇-水、乙腈-水3个不同比例为检测体系,结果发现目标峰出峰较晚,甚至不出峰,而甲醇-水(98∶2)首个目标峰出峰时间接近1 h,用乙腈替换也没明显改善出峰时间,而用纯甲醇作为流动相时,两个目标峰羽扇豆醇、羽扇豆酮的出峰时间明显缩短且分离效果好,故选择了纯甲醇作为流动相。
3.2 样品处理方法的选择 实验考察了不同溶剂(甲醇、乙醇、氯仿及氯仿-甲醇)作为提取溶剂,结果发现甲醇作为提取溶剂提取率相对较高,氯仿-甲醇(1∶1)及氯仿次之,乙醇作为提取溶剂时目标峰分离效果不好,故选用甲醇作为提取溶剂,同时本实验也考察了不同浓度甲醇(100%、90%、80%、75%、65%),不同提取方法(超声提取法、回流提取法、冷浸提取法),不同提取时间(0 min、30 min、40 min、50 min、60 min),不同料液比(15倍、25倍、35倍、45倍体积),最终确定以100%甲醇为提取溶剂,超声提取30 min,料液比为25倍时样品提取率相对较高且操作简便,故为最优处理方法。
3.3 样品含量测定 由表2可知,钝药野木瓜全株植物中的羽扇豆醇含量均高于纯茎部位,可能该植物的叶子含有较大量的羽扇豆醇成分,而羽扇豆酮的含量受植株部位的影响较小;由表3中结果显示,羽扇豆醇的含量受植物生长环境的影响较小,但不同产地还是存在差异,贵州丹寨地区的钝药野木瓜中羽扇豆醇含量最高,为0.0113 mg/g;羽扇豆酮的含量差异较大,植株受生长环境因素影响较大,并且贵州贵定地区的钝药野木瓜中羽扇豆酮与总三萜的含量最高,分别为0.0708 mg/g和0.0794 mg/g。贵州省8个不同产地、10批次的药材中,丹寨和贵定地区样品的羽扇豆醇和羽扇豆酮含量最高,分别为0.0113 mg·g-1和0.0708 mg·g-1。
实验首次从钝药野木瓜中分离、纯化和鉴定出4种五环三萜化合物,并建立HPLC法同时测定钝药野木瓜药材中羽扇豆醇和羽扇豆酮的含量,分析方法简单、可行,研究结果为钝药野木瓜药材的开发利用提供科学依据。
[1]国家中医药管理局. 中华本草 [M]. 上海:上海科学技术出版社, 1999:1936.
[2]贵州省药品监督管理局.贵州省中药材、民族药材质量标准[M]. 贵州:贵州科技出版社,2003:334.
[3]Wei Y., Ma C.M., Chen D.Y. et al. Anti-HIV-1 Protease Triterpenes from Stauntonia obovatifoliola Hayata subsp.intermedia. [J]. Phytochemistry, 2008(69):1875.
[4]国家药典委员会.中华人民共和国药典 [M]. 北京:中国医药科技出版社,2015: 314.
[5]胡冬群,杜江,李扬,等. 贵州钝药野木瓜药材的质量测定[J].贵州农业科学,2016, 44(4), 123.
[6]余良忠,文萍,虞金宝,等.HPLC法测定野木瓜中木犀草素的含量[J].中国实验方剂学,2013,19(9):111.
[7]余良忠,文萍,虞金宝,等.高效液相色谱法测定野木瓜中绿原酸的含量[J].医药导报,2008,27(7):854.
[8]唐唯媛,张义明,董永刚.HPLC法同时测定野木瓜中齐墩果酸与熊果酸的研究[J].中国食品添加剂(试验研究),2010(2):196-200.
[9]王淮滨,于德泉,梁晓天,等.野木瓜甙YM10和YM12的结构[J].药学学报,1989(6):444.
[10]彭小冰,危英,高伟略, 等. 尾叶那藤地上部分化学成分研究[J].中药材, 2013,36(11): 1796.
[11]陈超,李振兴,孟大利, 等.黄腊果藤茎化学成分的分离与鉴定[J].沈阳大学学报,2011,28(6):417.
[12]张琳,张有成. 三萜类化合物羽扇豆醇的抗肿瘤作用[J].国际肿瘤学杂志,2012, 39(2):114.
[13]Madureira AM,Ascenso JR, et al. Evaluation of the antiviral and antimicrobial activities of triterpenes isolated from Euphorbia segetalis[J] .Nat Prod Res,2003,17 (5): 375.
[14]Dantanarayana A P, Kumar N S, Muthukuda P M, et al. A lupine derivative and the 13 C NMR chemical shifts of some lupanone from Pleurostylia opposita [J]. Phytochemistry, 1982(21): 2065.
[15]Ikuta A, The triterpenes from Stauntonia hexaphylla callustissues and their Biosynthetic significance [J]. J. Nat. Prod. 1989(52): 623.
[16]Wenkert E, Baddeley G V, Burfit I R, et al. Carbon-13 nuclear magnetic resonance spectroscopy of naturally-occurring substances. LVII. Triterpenes related to lupane and hopane [J]. Org.Magn. Reson. 1978(11): 343.
[17]Ikuta, A., Itokawa, H., Triterpenoids of Paeonia japonica callus tissue [J]. Phytochemistry, 1988(27): 2813.
