PI3K/Akt/mTOR信号通路介导的N822K突变对c-KIT抑制剂诱导的AML细胞凋亡的影响①
2018-03-23徐建萍李国平傅晓萌郑金源林东红
徐建萍 李国平 吴 玮 傅晓萌 郑金源 李 丹 林东红
(福建医科大学医学技术与工程学院,福州 350004)
c-KIT,属于Ⅲ型酪氨酸激酶受体家族,位于人类染色体4q11~12上,基因序列高度保守,其mRNA大约5 084 bp。c-KIT作为干细胞因子的受体,可通过一系列信号通路参与造血干细胞增殖分化的调控[1]。近年来研究发现c-KIT基因突变,特别是激活性突变是导致以染色体 t(8;21)及 inv16为特征的核心结合因子急性髓系白血病(Core binding factor-acute myeloid leukemia,CBF-AML)患者预后不良的主要因素[2,3]。这些突变主要存在于8 和17号外显子,其中17号外显子酪氨酸激酶结构域的激活环(A-loop)中的D816V和N822K突变频繁,并与CBF-AML不良预后密切相关[4-6]。目前国内外对A-loop环的机制研究主要集中在c-KIT D816V突变[7,8],而关于N822K突变的研究则鲜见报道,已有的研究报道也主要集中在临床样本突变检出率及预后相关性[6,9,10],其在CBF-AML中的生物学作用和可能机制尚不明确。
基于此,本课题组前期开展了c-KIT N822K突变对AML细胞生物学影响的研究,发现在AML中位于A-loop环的N822K突变可使c-KIT发生干细胞因子非依赖性结构性激活;N822K突变的AML细胞对多靶点的受体酪氨酸激酶抑制剂舒尼替尼(Sunitinib)更敏感,较低浓度的舒尼替尼即可抑制其克隆形成和发生G0/G1期阻滞;然而流式凋亡检测结果却显示N822K突变AML细胞的凋亡率远低于无N822K突变的AML细胞,但其相关的凋亡分子机制尚不清楚,因此本实验进一步通过Western blot在蛋白水平研究c-KIT N822K突变对舒尼替尼诱导的AML细胞凋亡的影响,并探讨其相关的分子机制,以期为N822K突变作为干预CBF-AML治疗预后靶点的临床应用提供更多的实验依据。
1 材料与方法
1.1材料
1.1.1主要试剂 舒尼替尼购自美国Pfizer公司,β-actin抗体购自Bioworld公司,Bcl-2、Bax、c-myc、p-mTOR抗体购自Abcam公司,Actived-Caspase-3、PARP、Akt、PI3K抗体购自美国Cell Signaling Technology公司,Cyto C、Caspase-9抗体购自碧云天生物技术研究所,p-Akt、4EBP1、p-4EBP1、p-PI3K、mTOR抗体购自美国Signalway Antibody公司。
1.1.2主要仪器及器材 超净工作台购自香港Heal Force公司,低温高速台式离心机购自美国Thermo Fisher公司,低温冰箱购自美国Thermo Fisher公司,电泳槽、电泳仪、转膜装置(DYC40C)购自美国Bio-Rad 公司,微量加样器购自Eppendorf公司。
1.1.3细胞来源与培养 Kasumi-1细胞为复旦大学附属上海市第五人民医院血液科刘立根教授惠赠、HL-60、NB4细胞为福建省血液病研究所提供。经测序确定为c-KIT N822K突变型的急性髓系白血病Kasumi-1细胞作为实验组,用含20%Hyclone胎牛血清的RPMI1640培养液培养;经测序确定未发生c-KIT N822K突变的急性髓系白血病HL-60、NB4细胞作为对照组,分别用含15%和10%天津灏洋生物有限公司胎牛血清的RPMI1640培养液培养。均置于37℃,5%CO2饱和湿度的培养箱中培养,2~3 d 换液一次,取对数生长期的细胞进行实验。
1.2方法
1.2.1干预处理 取对数生长期的各株细胞,用0、0.04、0.16、0.64 μmol/L的舒尼替尼分别与细胞共同孵育24 h。其中,舒尼替尼浓度组包括:0.04、0.16、0.64 μmol/L的药物浓度组;0 μmol/L为未加药对照组。各浓度组种2瓶,15 ml/瓶,以保证收集到足够的细胞数以供后续实验。
1.2.2Western blot检测细胞中蛋白的表达 收集各组细胞,约5×105个/组,PBS洗涤细胞2次,加入RIPA裂解液裂解细胞,提取总蛋白,BCA法定量。Western blot操作方法见文献[11]。采用 Gel-Pro analyzer 软件系统对胶片进行图像扫描和分析,目的蛋白的相对表达量为目的条带与β-actin的灰度值之比。实验独立重复3次。
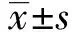
2 结果
2.1凋亡相关蛋白Western blot检测结果 用0、0.04、0.16、0.64 μmol/L的舒尼替尼分别与Kasumi-1、HL-60及NB4细胞共同孵育24 h后,检测线粒体参与的内源性凋亡通路。如图1、2所示,随着舒尼替尼浓度的增加,HL-60及NB4细胞均出现促凋亡蛋白Bax、Cyto C、Caspase-9、Actived-Caspase-3、PARP上调(P<0.05),抗凋亡蛋白 Bcl-2下调(P<0.01),基本都在0.04 μmol/L浓度组就开始变化。但在具有c-KIT N822K突变的Kasumi-1细胞中这些蛋白的增高趋势则明显减弱,Bax、Cyto C、Caspase-9等蛋白直到最高浓度0.64 μmol/L组才开始出现较明显的改变(P<0.05)。HL-60及NB4细胞中凋亡最终执行者Actived-Caspase-3蛋白及其作用底物PARP的89 kD剪接体也出现明显上调(P<0.05);但Kasumi-1细胞0.64 μmol/L组,PARP的89 kD剪接体仍无明显变化(P>0.05)。同时检测的原癌基因c-myc 蛋白可发现,Kasumi-1细胞c-myc蛋白在0.04 μmol/L浓度组出现下调(P<0.01),具有明显的剂量依赖性;而对照组HL-60和NB4细胞只在最高浓度0.64 μmol/L出现明显下调(P<0.01)。
2.2PI3K/Akt/mTOR通路Western blot检测结果 用0、0.04、0.16、0.64 μmol/L的舒尼替尼分别与Kasumi-1、HL-60及NB4细胞共同孵育24 h后,检测PI3K/Akt/mTOR通路蛋白。如图3、4所示,随着舒尼替尼浓度的增加,具有c-KIT N822K突变的Kasumi-1细胞PI3K/Akt通路的磷酸化蛋白p-PI3K、p-Akt、p-4EBP1在其各自总蛋白中的比例与0 μmol/L 浓度组有明显差异(P<0.05),出现典型的剂量依赖性下调,尤其是p-PI3K和p-Akt 蛋白在0.04 μmol/L浓度组就开始下调(P<0.01);但对照组HL-60及NB4细胞随着舒尼替尼浓度的增加其p-PI3K、p-4EBP1的表达与0 μmol/L浓度组无明显变化(P>0.05),p-Akt 蛋白在0.64 μmol/L浓度上调(P<0.05)。此外,在对此通路的重要下游分子mTOR检测时发现,具有c-KIT N822K突变的Kasumi-1细胞随着舒尼替尼浓度的增大,mTOR蛋白磷酸化的水平明显下调(P<0.01);而对照组HL-60和NB4细胞的mTOR蛋白磷酸化水平无明显变化(P>0.05)。
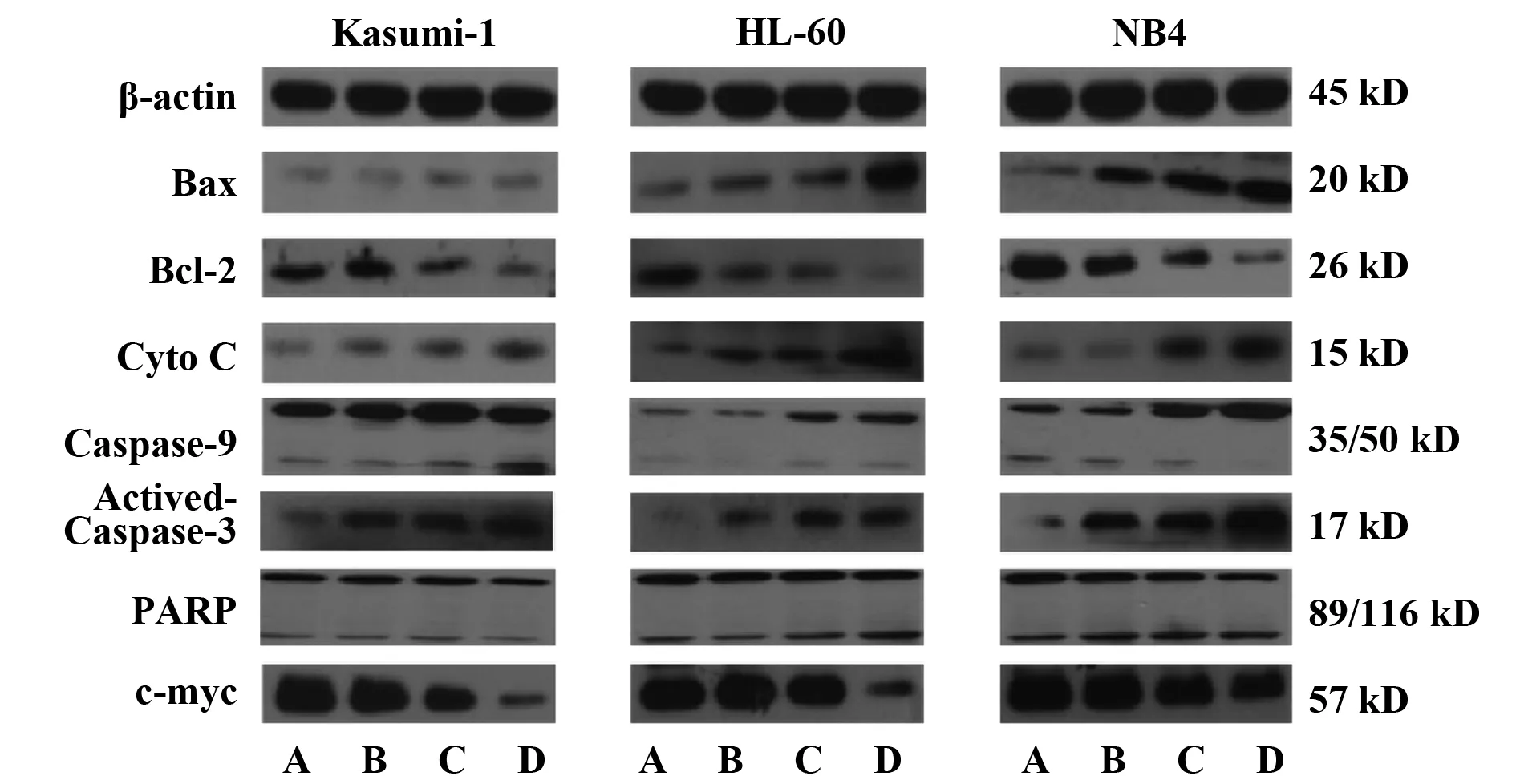
图1 不同浓度舒尼替尼作用24 h后三株细胞凋亡相关蛋白的表达Fig.1 Expression of apoptosis-related proteins of three cells treated by different concentrations of Sunitinib for 24 h Note: A.0 μmol/L;B.0.04 μmol/L;C.0.16 μmol/L;D.0.64 μmol/L.
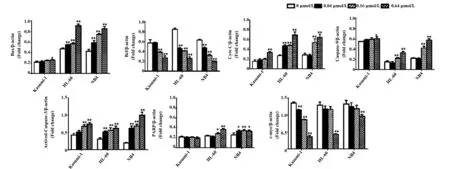
图2 不同浓度舒尼替尼作用24 h后三株细胞凋亡相关蛋白的相对表达量Fig.2 Relative expression of apoptosis-related proteins of three cells treated by different concentrations of Sunitinib for 24 hNote: *.P<0.05,**.P<0.01.
图3 不同浓度舒尼替尼作用24 h后三株细胞PI3K/Akt/mTOR通路蛋白的表达Fig.3 Expression of PI3K/Akt/mTOR proteins of three cells treated by different concentrations of Sunitinib for 24 hNote: A.0 μmol/L;B.0.04 μmol/L; C.0.16 μmol/L;D.0.64 μmol/L.
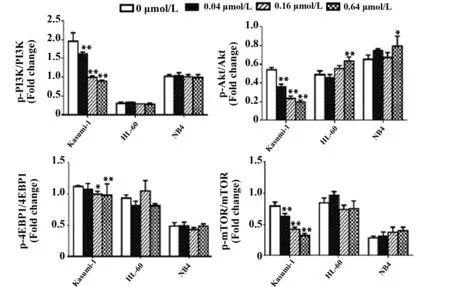
图4 不同浓度舒尼替尼作用24 h后三株细胞PI3K/Akt/mTOR通路蛋白的相对表达量Fig.4 Relative expression of PI3K/Akt/mTOR proteins of three cells treated by different concentrations of Sunitinib for 24 hNote: *.P<0.05;**.P<0.01.
3 讨论
CBF-AML是一群特殊的FAB分型为 M2 和 M4Eo 亚型的AML亚群,占 AML 患者5%~8%,其主要特征是8号和21号染色体异位t(8;21)(q22;q22)和16号染色体倒位inv(16) (p13q22)/t(16,16) (p13;q22)[12]。细胞遗传学预后分组中,CBF-AML被认为是预后较好的一种白血病类型[13]。
近年大量研究发现,c-KIT基因突变是导致CBF-AML 患者预后不良的主要因素[2,3]。c-KIT突变在随机急性髓系白血病(AML)案例中出现的频率只有2%~6.9%,但在高频率的CBF-AML中,高达48%[14-16]。在所有c-KIT突变中,TK2区域尤其是A-loop环处的突变检出频率明显高于其他区域,而A-loop环中又以D816V和N822K突变与总生存期(Overall survival,OS)有显著负相关[5]且突变率高。美国 NCCN(National Compre-hensive Cancer Network)指南已将D816V突变列为AML的重要预后指标。随着研究的深入,N822K突变也因其可导致AML 患者复发率高、治愈率低引起了人们的关注[9]。
众所周知,Kasumi-1细胞是从M2亚型的AML患者外周血中分离建株的[17],并且具有c-KIT第822个密码子(N822K)的点突变[18],可作为CBF-AML和c-KIT N822K突变研究的最合适的细胞模型。Beghini等[19]研究发现,酪氨酸激酶抑制剂——甲磺酸伊马替尼(imatinib mesilate,商品名:格列卫),可抑制c-KIT活性,诱导增殖抑制和具有c-KIT N822K突变的细胞凋亡。但其在包括Kasumi-1在内的AML细胞内药物积累量低于舒尼替尼,故舒尼替尼敏感性更高[20]。Ikezo等[21]筛查各种恶性血液病细胞时也发现舒尼替尼对于具有酪氨酸激酶激活突变的白血病细胞,包括EOL-1、MV4-11和Kasumi-1细胞有很强的诱导凋亡能力。因此,本课题组前期通过17外显子测序确认具有N822K突变的Kasumi-1细胞作为实验组,选择经确认无N822K突变的HL-60和NB4作为对照细胞,选取舒尼替尼作为c-KIT抑制剂,研究N822K突变对c-KIT抑制剂诱导AML细胞凋亡的影响,其对生物学特性影响的相关结果已总结论文待发表。其中,流式凋亡检测结果显示,随着舒尼替尼浓度从0 μmol/L增加到0.80 μmol/L,Kasumi-1细胞的凋亡率从(19.41±1.83)%上升到(33.30±4.36)%(P<0.05),而无N822K突变的HL-60和NB4细胞其凋亡率分别上升到87.85% 和43.97%。可见舒尼替尼抑制c-KIT活性后,具有N822K突变AML细胞的凋亡率明显低于无N822K突变的AML细胞,因此,本研究拟进一步探讨其相关的凋亡分子机制。
我们首先检测了caspase通路的凋亡相关蛋白,结果显示随着舒尼替尼浓度的增加,对照组HL-60及NB4细胞出现线粒体参与的内源性凋亡通路蛋白的典型变化:Bcl-2下调及Bax上调促进Cyto C的释放,Caspase-9被活化,进而激活细胞凋亡最终执行者Caspase-3,剪切底物PARP,最终导致DNA被裂解,细胞凋亡;而具有c-KIT N822K突变的Kasumi-1细胞这种趋势变化则明显减弱。同时检测可促进细胞从G0/G1期进入S期的c-myc 蛋白时发现,Kasumi-1细胞原癌基因c-myc出现了明显的剂量依赖性下调,而对照组HL-60和NB4细胞只在最高浓度0.64 μmol/L组出现明显下调。以上结果与我们前期对生物学特性改变的研究结果一致,进一步表明可能由于N822K突变诱导的结构性激活的存在,使Kasumi-1细胞对舒尼替尼凋亡诱导作用及增殖抑制的反应不同于无N822K突变的HL-60及NB4细胞。
有研究表明,c-KIT D816v突变最明显的通路改变是PI3K/Akt通路,该通路主要作用是抑制细胞凋亡、促进细胞周期进展,从而促进细胞的生存和增殖[22]。因此我们设想,在c-KIT N822K突变的AML细胞中也存在PI3K/Akt通路的变化,并影响舒尼替尼对其凋亡诱导作用。通过进行相关实验,结果发现,随着舒尼替尼浓度的增加,具有c-KIT N822K突变的Kasumi-1细胞出现了磷酸化p-PI3K、p-Akt、p-4EBP1剂量依赖性下调;但对照组HL-60及NB4细胞对PI3K/Akt通路蛋白的抑制作用则较弱。分析其原因可能是:Kasumi-1细胞具有N822K突变而出现c-KIT结构性激活,细胞内PI3K/Akt通路处于高活性状态,因而对舒尼替尼的酪氨酸激酶抑制作用尤其敏感;而无c-KIT结构性激活作用的对照组HL-60及NB4细胞,细胞内PI3K/Akt通路本身为低活性,故对较低浓度的抑制剂不敏感,PI3K/Akt通路磷酸化蛋白下调现象不明显。
尤其值得一提的是,在本实验中发现,作为PI3K/Akt通路一个重要的下游分子mTOR,具有c-KIT N822K突变的Kasumi-1细胞随着舒尼替尼浓度的增大,mTOR分子磷酸化蛋白被明显抑制,其促进细胞从G0/G1期向S期转换的能力也可能因此被削弱,而出现舒尼替尼剂量依赖性的G0/G1期阻滞;而对照组的HL-60和NB4细胞mTOR磷酸化蛋白抑制作用不明显,G0/G1期阻滞作用也不明显。
综上所述,舒尼替尼对具有c-KIT N822K突变的AML细胞的G0/G1周期阻滞和增殖抑制作用可能是通过c-myc 蛋白和mTOR分子共同发挥作用的;抑制N822K突变引起的c-KIT结构性激活可能经抑制PI3K/Akt/mTOR通路,而削弱舒尼替尼对Kasumi-1细胞的凋亡诱导作用。
本课题组前期研究表明,在舒尼替尼作用下,Kasumi-1细胞出现明显增殖抑制和周期阻滞与其低凋亡率不符,因此我们思考除凋亡外,N822K突变的Kasumi-1细胞是否还通过其他方式死亡?本研究通过对凋亡相关蛋白和PI3K/Akt/mTOR通路蛋白水平检测发现舒尼替尼作用后的Kasumi-1细胞出现明显的mTOR抑制。Willems等[23]最近的研究成果引起了我们的高度关注,其在研究移植AML细胞的小鼠模型中发现,抑制mTOR可诱导细胞发生自噬,在此过程中有显著的自噬和部分凋亡发生,小鼠存活率明显升高。那么,我们用舒尼替尼作用后的Kasumi-1细胞是否也是发生了自噬呢?有待进一步深入研究。
[1] Ashman LK,Griffith R.Therapeutic targeting of c-KIT in cancer[J].Exp Opinion Invest Drugs,2013,22(1):103-115.
[2] Dombret H,Preudhomme C,Boissel N.Core binding factor acute myeloid leukemia (CBF-AML): is high-dose Ara-C (HDAC) consolidation as effective as you think?[J].Curr Opinion Hematol,2009,16(2):92-97.
[3] Riera L,Marmont F,Toppino D,etal.Core binding factor acute myeloid leukaemia and c-KIT mutations[J].Oncol Rep,2013,29(5):1867-1872.
[4] Kim HJ,Ahn HK,Jung CW,etal.KIT D816 mutation associates with adverse outcomes in core binding factor acute myeloid leukemia,especially in the subgroup with RUNX1/RUNX1T1 rearrangement[J].Annals Hematol,2013,92(2): 163-171.
[5] Wang D,Qiao C,Xiao M,etal.Integrative analysis of prognostic factors in Chinese core binding factor leukemia[J].Biochem Biophys Res Commun,2012,428(3):411-415.
[6] 符 爽,胡延平,陈 芳,等.伴ETO或CBFβ阳性的急性髓细胞白血病患者c-kit基因突变的检测及意义[J].现代肿瘤医学,2015,23(11):1581-1584.
Fu S,Hu YP,Chen F,etal.Detection of c-kit mutations in acute myeloid leukemia patients with ETO or CBFβ positive and its clinical significance[J].Modern Oncol,2015,23(11):1581-1584.
[7] Nick HJ,Kim HG,Chang CW,etal.Distinct classes of c-Kit-activating mutations differ in their ability to promote RUNX1-ETO-associated acute myeloid leukemia[J].Blood,2012,119(6): 1522-1531.
[8] Houcine B,Sophie GL,Gandhi D,etal.Relocalization of KIT D816V to cell surface after dasatinib treatment: potential clinical implications[J].Clin Lymphoma,Myeloma Leukemia,2013,13 (1):62-69.
[9] Shimada A,Taki T,Kubota C,etal.N822 mutation of KIT gene was frequent in pediatric acute myeloid leukemia patients with t (8;21)in Japan:a study of the Japanese childhood AML cooperative study group[J].Leukemia,2007,21(10):2218-2219.
[10] Yui S,Kurosawa S,Yamaguchi H,etal.D816 mutation of the KIT gene in core binding factor acute myeloid leukemia is associated with poorer prognosis than other KIT gene mutations[J].Annals Hematol,2017,96(10):1641-1652.
[11] 薛 龑,陈梅环,唐永金,等.SARI基因促进急性髓性白血病细胞凋亡机制的探讨[J].中国免疫学杂志,2017,33(11):1621-1625.
Xue Y,Chen MH,Tang YJ,etal.Mechanism of SARI gene promoting apoptosis in acute myeloid leukemia cells[J].Chin J Immunol,2017,33(11):1621-1625.
[12] Han LN,Zhou J,Schuringa JJ,etal.Treatment strategies in acute myeloid leukemia[J].Chin Med J,2011,124(9):1409-1421.
[13] Hyde RK,Liu PP.RUNX1 repression-independent mechanisms of leukemogenesis by fusion genes CBFB-MYH11 and AML1-ETO (RUNX1-RUNX1T1)[J].J Cell Biochem,2010,110(5):1039-1045.
[14] Beghini A,Peterlongo P,Ripamonti CB,etal.C-kit mutations in core binding factor leukemias[J].Blood,2000,95(2):726-727.
[15] Wang YY,Zhou GB,Yin T,etal.AML1-ETO and C-KIT mutation/Overexpression in t(8;21) leukemia: implication in stepwise leukemogenesis and response to Gleevec[J].Proc Natl Acad Sci U S A,2005,102(4):1104-1109.
[16] Fritschepolanz R,Fritz M,Huber A,etal.High frequency of concomitant mastocytosis in patients with acute myeloid leukemia exhibiting the transforming KIT mutation D816V[J].Mol Oncol,2010,4(4):335-346.
[17] Asou H,Tashiro S,Hamamoto K,etal.Establishment of a human acute myeloid leukemia cell line (Kasumi-1) with 8;21 chromosome translocation[J].Blood,1991,77(9):2031-2036.
[18] Larizza L,Magnani I,Beghini A.The Kasumi-1 cell line: a t(8;21)-kit mutant model for acute myeloid leukemia[J].Leukemia Lymphoma,2005,46(2):247-255.
[19] Beghini A,Megnani I,Ripemonti C,etal.Amplification of anovel C-Kit activation mutation Asn(822)-Lys in the Kasumi-1 cell line: a t(8;21)-C-Kit mutation for acute myeloid leukemia[J].Hematol J,2002,3(3):157-163.
[20] Hu S,Niu H,Minkin P,etal.Comparison of antitumor effects of multitargeted tyrosine kinase inhibitors in acute myelogenous leukemia[J].Mol Cancer Therapeutics,2008,7(5):1110-1120.
[21] Ikezoe T,Nishioka C,Tasaka T,etal.The antitumor effects of sunitinib (formerly SU11248) against a variety of human hematologic malignancies: enhancement of growth inhibition via inhibition of mammalian target of rapamycin signaling[J].Mol Cancer Therapeutics,2006,5(10):2522-2530.
[22] Timokhina I,Kissel H,Stella G,etal.KIT signaling through PI3-kinase and Src kinase pathways: an essential role for Rac1 and JNK activation in mast cell proliferation[J].EMBO J,1998,17(21):6250-6262.
[23] Willems L,Chapuiset N,Puissantal A,etal.The dual mTORC1 and mTORC2 inhibitor AZD8055 has anti-tumor activity in acute myeloid leukemia[J].Leukemia,2012,26(6):195-202.
