瑞士乳杆菌调控小鼠肠道菌群变化规律的研究
2018-01-08臧凯丽崔文静马新颖赵林森阎亚丽陈庆森
臧凯丽,贾 彦,崔文静,马新颖,王 泳,赵林森,赵 培,叶 雷,阎亚丽,*,陈庆森,*
(1.天津商业大学生物技术与食品科学学院,天津市食品生物技术重点实验室,天津 300134;2.河北一然生物科技有限公司,河北 石家庄 050899)
瑞士乳杆菌调控小鼠肠道菌群变化规律的研究
臧凯丽1,贾 彦1,崔文静2,马新颖2,王 泳1,赵林森2,赵 培1,叶 雷1,阎亚丽1,*,陈庆森1,*
(1.天津商业大学生物技术与食品科学学院,天津市食品生物技术重点实验室,天津 300134;2.河北一然生物科技有限公司,河北 石家庄 050899)
为探究瑞士乳杆菌(Lactobacillus helveticus)TS206对小鼠肠道菌群的调控规律,更好地阐述益生菌维持肠道菌群稳态的机制,本研究以雄性BALB/c小鼠为实验对象,将其分为L. helveticus TS206组、对照组、空白组。L. helveticus TS206组和对照组分别每天灌胃L. helveticus TS206菌悬液和等量的生理盐水,连续灌胃7 周,空白组不灌胃。每周采集小鼠新鲜粪便样品,通过试剂盒提取粪便细菌总DNA,利用Ion torrent个人化操作基因组测序平台测序技术对16S rRNA基因的V6区进行高通量测序,最后通过生物信息学和多变量统计学方法对测序数据进行分析。结果显示:所有测序序列在97%相似水平划分得到1 617 个操作分类单位(operational taxonomic units,OTUs),被划分为8 个门,硬壁菌门(Firmicutes)和拟杆菌门(Bacteroidetes)为各组小鼠中的优势菌门,占总序列数的97.49%,且硬壁菌门丰度最高,超过70%;优势菌科主要为S24-7、毛螺菌科(Lachnospiraceae)、瘤胃球菌科(Ruminococcaceae)、理研菌科(Rikenellaceae)和拟杆菌科(Bacteroidaceae),且与对照组相比L. helveticus TS206组肠杆菌科细菌数量较低并呈下降趋势。经LEfSe分析有53 个关键OTUs与2 组小鼠肠道粪便菌群结构显著相关,其中27 个OTUs在对照组中富集,分属于肠杆菌科、瘤胃球菌科、毛螺菌科和梭菌目;26 个OTUs在L. helveticus TS206组中富集,分属于梭菌目、毛螺菌科和拟杆菌属,其中与对照组相比,梭菌目丰度水平高。经主成分分析,两组样品完全分离,菌群的整体结构存在差异,且富集的OTUs之间呈负相关关系。综上所述,在实验阶段,灌胃雄性BALB/c小鼠一定剂量的L. helveticus TS206后,经扩增子测序分析获得的初步结论为L. helveticus TS206能改变肠道菌群整体结构的功效,表现出了抑制肠道有害微生物生长的作用,并通过促进部分有益菌的增殖来维持肠道菌群结构的稳态。
瑞士乳杆菌TS206;肠道菌群;Ion torrent个人化操作基因组测序技术;生物信息学;多变量统计学分析
Lederberg等[1]提出了人是一种由自身细胞和肠道微生物组成的“超级生物体”。人体肠道菌群编码的基因组可被视为“人的第二基因组”,人的基因组与人体肠道微生物宏基因组相互协调、和谐一致,共同参与人体的营养摄入、代谢和免疫的调控过程。到目前为止,众多的研究表明肠道菌群的结构和功能与机体的健康状况息息相关,肠道菌群影响宿主的代谢显型[2]、营养物质的产生和吸收[3]以及调整和改善机体的免疫系统[4]。肠道内菌群的任何动态平衡的打破都会影响胃肠道内的正常活动,造成肠道内的疾病,如炎性肠病、肠应激综合症、过敏性反应等[5],特别是与饮食不当造成的“现代文明病”如肥胖、糖尿病、心脑血管疾病、结肠癌等发生发展具有密切的关系[6-9]。因此,肠道菌群结构可以忠实而精细地反映人体的健康状况,而益生菌可以通过改变肠道内微生物结构,维持微生物区系的平衡来调控机体的代谢功能和肠黏膜屏障功能,这对肠道相关疾病的预防和治疗具有重要意义[10]。目前,国内外常用的益生菌菌种主要集中在功能特性比较清晰的乳杆菌和双歧杆菌属,如嗜酸乳杆菌(Lactobacillus acidophilus)、瑞士乳杆菌(Lactobacillus helveticus)、干酪乳杆菌(Lactobacillus casei)、植物乳杆菌(Lactobacillus plantarum)以及双歧杆菌(Bif i dobacterium)等[11-12]。
近年来,高通量测序技术是宏基因组学研究中应用最广泛的测序技术[13],主流的高通量测序技术主要包括454焦磷酸测序平台、美国Illumina公司的Hiseq和Miseq测序平台以及美国Life Technologies公司的Ion torrent个人化操作基因组测序仪(personal genome machine,PGM)和Ion proton测序平台。基于16S rRNA基因可变区高通量测序的技术(扩增子测序)被广泛用于肠道微生物群落结构分析中,为解析肠道菌群结构的变化与人体健康的关系提供了很好的技术平台。
到目前为止,就L. helveticus而言,国内外的研究主要集中在其发酵产生的生物活性物质(比如血管紧张素转化酶抑制肽等)对机体健康的促进作用。有关L. helveticus调控小鼠肠道菌群为主题的国内外文献及研究报道较少。王友湘等[14]采用选择性培养基探讨了L. helveticus TS206干预对小鼠肠道菌群结构的影响,发现小鼠肠道内乳酸菌、双歧杆菌、肠球菌等有益菌的数量显著增加,提示L. helveticus TS206能够促进小鼠肠道内有益菌的增殖,能够维持小鼠肠道微生态系统的平衡。而Frece等[15]通过传统的平板计数法检测小鼠粪便中相关细菌的数量,发现灌胃L. helveticus 8 d后小鼠肠道中乳酸杆菌数量增加而肠杆菌以及梭菌属细菌减少,这也与王友湘等[14]的研究结论相吻合。Taverniti等[16]指出,一些体外研究表明L. helveticus表现出许多常见的益生菌特性,如肠胃中生存能力、定植上皮细胞和抵抗病原体的能力,体内小鼠模型研究表明L. helveticus可以预防胃肠道感染,增强其对病原体的抵抗力,调节宿主免疫反应,并影响肠道菌群的组成。
鉴于针对L. helveticus的这类研究方法较少,所以本研究参考Singh等[17]给小鼠灌胃Bifidobacterium bifidum MIMBb75,利用定量聚合酶链式反应(polymerase chain reaction,PCR)技术检测小鼠肠道菌群变化的方法,以L. helveticus TS206为研究对象,利用扩增子测序技术对长期L. helveticus TS206干预小鼠的肠道菌群结构进行分析,探究L. helveticus TS206调控小鼠肠道菌群结构变化的规律并鉴定出与其干预显著相关的关键菌属。本研究将丰富L. helveticus TS206的益生功能,为该菌作为功能性食品改善人体健康提供可靠的理论依据,还为以肠道菌群为靶点通过益生菌的摄入来预防各类慢性疾病带来了全新的思路和方法。
1 材料与方法
1.1 材料与试剂
L. helveticus TS206,天津商业大学实验室保藏。
110 只6 周龄BALB/c雄性小鼠,SPF级,购自中国人民解放军军事医学科学院实验动物中心,许可证号SCXK(京)2009-0017。小鼠饲料为普通繁育饲料,SPF级,购于中国人民解放军军事医学科学院实验动物中心。
MRS液体培养基 北京奥博星生物技术有限责任公司;乳清粉液体培养基(质量分数10%的乳清粉液体培养基)、肠道菌计数琼脂(violet red bile dextrose agar,VRBDA)培养基 青岛海博生物有限公司;QIAamp Fast DNA Stool Mini Kit 德国QIAGEN企业;Pfu DNA聚合酶、Ion Plus Fragment Library Kit、Ion Xpress™ Barcode Adapters Kit、Ion PGM™Template OT2 200 Kit、Ion PGM™ Sequencing 200 Kit v2美国赛默飞世尔科技公司;MiniBEST Agarose Gel DNA Extraction Kit 宝生物工程(大连)有限公司;TAE缓冲液(50×) 上海生物工程有限公司;High Sensitivity DNA Kit 美国安捷伦公司; Qubit®dsDNA HS Assay Kit 美国Invitrogen公司;Agencourt®AMPure®XP Kit美国贝克曼公司。
1.2 仪器与设备
900 SERIES超低温冰箱 美国赛默飞世尔科技公司;SW-CJ-2DD单人双面净化工作台 苏州净化设备有限公司;3K18高速冷冻离心机 美国Sigma公司;DYY-2C电泳仪 北京六一仪器厂;Ion torrent个人化操作基因组测序平台、INS1005527模板制备系统(Ion One TouchTM2)、8441-21模板富集系统(Ion OneTouch™ ES)美国赛默飞世尔科技公司;331-1生物分析仪 美国安捷伦公司;N8050200荧光定量PCR仪 美国ABI公司;Qubit 2.0核酸蛋白定量仪、12321D磁力架 美国Invitrogen公司;II-3DNA紫外-可见分光光度计 英国柏诺公司。
1.3 方法
1.3.1 L. helveticus TS206的培养和富集
将保藏的L. helveticus TS206于乳清粉液体培养基40 ℃活化2 代。将活化后的L. helveticus TS206液按体积分数1%的接种量接种在MRS液体培养基中,40 ℃静置培养18 h至对数期,5 000 r/min 4 ℃离心10 min收集菌体细胞,无菌生理盐水洗涤2 次,最后用10 mL无菌生理盐水悬浮使菌体浓度为3×108~4×108CFU/mL,以备后续动物实验灌胃使用。
1.3.2 动物实验
1.3.2.1 动物分组与灌胃
110 只SPF级雄性BALB/c小鼠适应性喂养1 周后随机分为L. helveticus TS206组、对照组和空白组,其中L. helveticus TS206组45 只,对照组45 只,空白组20 只;L. helveticus TS206组小鼠每天灌胃0.2 mL L. helveticus TS206菌悬液,对照组小鼠每天灌胃等量的生理盐水,连续灌胃7 周,灌胃结束后再继续自由饮食饮水饲养1 周,空白组不灌胃;实验期间各组小鼠均饲喂普通繁育饲料,自由进食和饮水,同时对小鼠的毛发色泽、饮食状况、活动程度进行记录。
1.3.2.2 小鼠粪便采集
分别在实验开始后第0、1、2、3、4、5、6、7、8周早上8 点,采用逼迫法采集L. helveticus TS206组和对照组小鼠新鲜粪便,在第2、4、6、8周采集空白组小鼠粪便,并立即放入冰盒保存后带回实验室进行后续实验。
1.3.3 Ion torrent PGM测序技术探究L. helveticus TS206对小鼠肠道菌群结构的影响
1.3.3.1 小鼠粪便中细菌基因组DNA的提取
迅速称取-80 ℃冻存的1.3.2.2节所采集小鼠粪便样品0.2 g,按照QIAamp Fast DNA Stool Mini Kit的使用说明提取细菌基因组DNA。用DNA紫外-可见分光光度计进行核酸质量浓度检测,并用0.8%琼脂糖凝胶电泳检测基因组DNA的完整性。
1.3.3.2 肠道细菌16S rRNA V6区的PCR扩增
利用细菌通用引物扩增1 6 S r R N A V 6区片段,上游引物为以下两引物的等量混合:9 6 7 F(5’-CAACGCGAAGAACCTTACC-3’)和967F(5’-ATACGCGAGGAACCTTACC-3’),下游引物为1046R(5’-CGACARCCATGCASCACCT-3’)。PCR反应体系(50 μL):5 μL缓冲液(10×)(无Mg2+)、3 μL 25 mmol/L MgSO4、1 μL 10 mmol/L的dNTP、上下游引物(10 μmol/L)各0.5 μL,50 ng肠道细菌基因组模板DNA,最后加入0.4 μL Pfu DNA 聚合酶(2.5 U/μL),加无菌水至50 μL。为避免非特异性扩增采用降落PCR的方法进行扩增,反应程序:95 ℃预变性3 min;95 ℃变性1 min;退火温度从65~55 ℃,每个循环降0.5 ℃,55 ℃ 15 个循环,退火30 s;72 ℃延伸1 min,一共35 个循环;最后72 ℃延伸7 min。PCR产物用3%的琼脂糖凝胶电泳检测并用MiniBEST Agarose Gel DNA Extraction Kit纯化。纯化后的扩增产物用Qubit®dsDNA HS Assay Kit进行PCR产物的定量。
1.3.3.3 Ion torrent测序
先用Ion Plus Fragment Library Kit和Ion Xpress™Barcode Adapters Kit进行测序文库的构建。然后进行测序模板的制备,Ion OneTouch™ ES仪器自动对携带有测序模板的Ion Sphere™ Particles进行富集。最后在Ion torrent个人化操作基因组测序平台上进行上机测序。
1.3.3.4 测序数据生物信息学和多变量统计学分析
原始数据利用Fast QC软件进行质控,并利用NG Stoolkits过滤掉低质量序列,利用Usearch方法对所得的高质量序列,根据样品条码进行样品的分选,在97%的相似水平下划分分类操作单元(operational taxonomic units,OTUs),生成以序列数代表的每个样本中每个OTUs丰度的OTU Table(biom文件)。然后按照微生物生态学定量分析(quantitative insights into microbial ecology,QIIME)[21]的流程进行物种分类地位的确定,并进行稀疏曲线的绘制,计算观测到的物种(observed species)指数和Shannon指数。最后,计算结果用R语言
3.1.3 软件进行作图。基于上述得到的OTU Table应用基于线性判别分析(linear discriminant analysis,LDA)的LEfSe(linear discriminant analysis effect size)算法筛选组间差异OTUs。根据这些差异OTUs的丰度利用R语言3.1.3软件进行丰度聚类热图的绘制和斯皮尔曼相关性系数的计算,并利用Cytoscape 3.2.1软件绘制互作网络图。
1.4 数据统计分析
采用了R语言软件的wilcox检验对数据进行了显著性分析,P<0.05表示差异显著。
2 结果与分析
2.1 L. helveticus TS206干预后小鼠肠道微生物整体特征
2.1.1 各样品中序列和OTUs数目概况
22 个样本中除去2 个误差较大的样本,余下的20 个样本共获得3 001 460 条高质量序列,每个样品的序列分布情况如表1所示。全部序列的长度均在80~120 bp的范围内,由于细菌16S rRNA基因的V6区平均长度为100 bp,所以本次实验的测序长度基本能覆盖V6区。

表1 各样品中序列和OTUs(97%相似水平)分布Table 1 Numbers of reads and OTUs (97% similarity level) in the library of each sample
在本实验中,根据16S rRNA基因序列相似度与细菌分类地位之间的对应关系,在97%的序列相似性水平上经过Usearch的方法划分OTUs。每个样本的OTUs分布如表1所示,20 个样本一共获得了1 617 个OTUs。
2.1.2 L. helveticus TS206干预后小鼠肠道微生物整体结构的改变
通过97%相似水平划分得到1 617 个OTUs,OTUs的代表序列与Greengene 1308软件的数据库进行比对注释得到每个OTU的物种注释情况,最后统计各个样本中各分类水平相对丰度,结果如图1所示。

图1 L. helveticus TS206对小鼠肠道菌群门水平上相对丰度的影响Fig. 1 Effect of L. helveticus TS206 on relative abundance of dominant bacterial phyla in the intestine of mice
图1显示所有OTUs被划分为8 个门,其中硬壁菌门(Firmicutes)和拟杆菌门(Bacteroidetes)为各组小鼠粪便中的优势菌门,占总序列数的97.49%。其中硬壁菌门的含量最丰富,约占总序列数的70.66%,含有1 147 个OTUs,其中99.3%的序列属于Clostridia纲,含有1 135 个OTUs。而拟杆菌门则占总序列数的26.83%,含有313 个OTUs全部属于Bacteroidales目,其中54.9%的序列是S24-7科细菌,分属177 个OTUs;其次是拟杆菌科(Bacteroidaceae)占17.8%,分属37 个OTUs;理研菌科(Rikenellaceae)含有36 个OTUs,占11%;其他序列分属于普雷沃氏菌科(Prevotellaceae)、Odoribacteraceae科、Paraprevotellaceae科和紫单孢菌科(Porphyromonadaceae),含有63 个OTUs。其余的如放线菌门(Actinobacteria)3 个OTUs、蓝细菌门(Cyanobacteria)9 个OTUs、脱铁杆菌门(Deferribacteres)1 个OTU、变形菌门(Proteobacteria)17 个OTUs、无壁菌门(Tenericutes)59 个OTUs和疣微菌门(Verrucomicrobia)3 个OTUs,它们的相对丰度较低,在各样品中所占比例基本都不足1%,另外,还有0.5%左右的无法确定具体分类地位的细菌。
从图1可以看到,L. helveticus TS206灌胃组小鼠肠道拟杆菌门(Bacteroidetes)细菌的相对丰度随着灌胃L. helveticus TS206时间延长有降低的趋势,在灌胃第6周拟杆菌门细菌相对丰度降到最低,由最初的27.5%降到17.8%;对照组小鼠肠道拟杆菌门细菌丰度虽然在第2、3周(B2、B3)有所波动,但实验期间基本维持在同一水平。伴随着拟杆菌门细菌相对丰度的降低,L. helveticus TS206灌胃组小鼠肠道硬壁菌门细菌相对丰度相应升高,由起初的71.4%增加到第6周的80.7%。

图2 L. helveticus TS206对小鼠肠道菌群科水平上相对丰度的影响Fig. 2 Effect of L. helveticus TS206 on relative abundance of major bacterial families in the intestine of mice
各组小鼠肠道菌群在科水平上主要物种相对丰度的变化如图2所示。在科的水平上丰度占优势地位的主要为S24-7、毛螺菌科(Lachnospiraceae)、瘤胃球菌科(Ruminococcaceae)、拟杆菌科(Bacteroidaceae)和理研菌科(Rikenellaceae)的细菌。经过干预后,对瘤胃球菌科细菌作用持续性较好,对动物的生理影响也比较持久。而毛螺菌科细菌在灌胃L. helveticus TS206后相对丰度波动较大,但是总体上都高于对照组。同时,在灌胃L. helveticus TS206后有显著变化的理研菌科从起初的2.9%降低到第7周(A7)的0.7%,也显著低于对照组(4.1%);S24-7科细菌从最初的27.5%降低到第6周(A6)的11.3%,也低于对照组的平均水平(17.4%)。综合在门水平上的比较结果,可以得知灌胃L. helveticus TS206后小鼠肠道菌群拟杆菌门细菌丰度的降低可能是由于理研菌科和S24-7科细菌的降低引起的。
另外,对各组小鼠肠道中丰度较低的肠杆菌科(Enterobacteriaceae)细菌的相对丰度也进行了比较,发现虽然它们在各组小鼠肠道中的总体相对丰度较低,且在各个样本中的个体差异也比较大,但是总体而言在L. helveticus TS206组小鼠肠道中肠杆菌科细菌的相对丰度要比对照组低一个数量级。这一结果通过传统的平板计数的方法得到了验证,结果如图3所示。在整个实验过程中,L. helveticus TS206组小鼠肠道中肠菌科细菌数量具有降低趋势,到灌胃第4周(A4)降到最低,相比于对照组,随后一直处于较低水平;而对照组小鼠肠杆菌科细菌则是随着实验时间的延长而增加。虽然在L. helveticus TS206灌胃前两周时,对照组小鼠肠杆菌数量都低于L. helveticus TS206组,差异极显著(P<0.01),但是从第3周开始一直到实验结束,L. helveticus TS206灌胃组小鼠肠道内肠杆菌数量一直显著地低于对照组(P<0.05)。

图3 瑞士乳杆菌对小鼠肠道中肠杆菌科细菌数量的影响Fig. 3 Effect of L. helveticus TS206 on the number of intestinal Enterobacteriaceae in mice
2.2 L. helveticus TS206对小鼠肠道微生物α多样性的影响
为了进一步研究L. helveticus TS206对小鼠肠道微生物整体结构的影响,从各样本肠道菌群的丰富度和均匀度的层面研究小鼠肠道微生物的多样性。以各样品在97%相似水平上划分的OTUs数据为对象,通过QIIME分析了各样本的α多样性,首先通过稀释曲线和反映样本物种丰富度的observed species指数来研究两组样本物种多样性的差异,其次通过反应微生物多样性和均匀度的Shannon指数来研究两组样本的多样性和均匀度的差异,发现L. helveticus TS206对小鼠肠道微生物α多样性造成了一定程度的影响。

图4 对照组和益生菌组各样本稀释曲线Fig. 4 Rarefaction curves of OUT diversity for control and probiotic groups
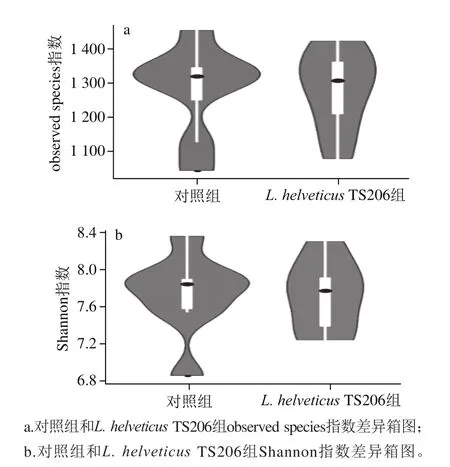
图5 L. helveticus TS206对肠道微生物α多样性指数的影响Fig. 5 Effect of L. helveticus TS206 on α diversity index of gut microbiota
图4结果表明各组样本的OTUs数目随着测序深度的增加基本达到饱和,说明当前测序深度足以发现各样本生境中的大部分物种。而2 组样本的稀释曲线交错,说明2 组小鼠肠道物种多样性并没有大的差异;这与图5a两组小鼠肠道微生物observed species指数差异分析箱图的结果一致,虽然差异分析箱图显示对照组要略高于L. helveticus TS206组,但差异不显著(P>0.05)。
图5b中两组小鼠肠道微生物Shannon指数的差异分析也出现同样结果,在灌胃L. helveticus TS206后α多样性指数与对照组相比较低,但是差异不显著(P>0.05),以上说明L. helveticus TS206对小鼠肠道微生物整体多样性没有显著影响(P>0.05)。
2.3 与2 组小鼠肠道粪便菌群结构差异显著相关的肠道关键OTUs
上述肠道微生物整体结构的分析发现,2 组小鼠肠道微生物具有一定程度的差异。采用LEfSe差异分析算法筛选出L. helveticus TS206组与对照组间具有显著差异的OTUs,结果如图6所示。

图6 与L. helveticus TS206灌胃相关的肠道关键OTUsFig. 6 Key OTUs of gut microbiota responding to L. helveticus TS206
通过LEfSe分析寻找到53 个OTUs是造成L. helveticus TS206组和对照组小鼠肠道菌群结构差异的关键物种。相对于对照组,27 个OTUs在L. helveticus TS206组中丰度较低,26 个OTUs在L. helveticus TS206组丰度富集(图6a)。
从图6b中可以发现,共有3 个OTUs(OTU1 488、OTU1 588、OTU1 598)属于肠杆菌科和1 个属于瘤胃球菌科的OTUs均在对照组中富集;同时,在这53 个差异OTUs中,有15 个OTUs属于梭菌目(Clostridiales),而7 个属于梭菌目的OTUs以及部分属于拟杆菌属的OTUs在L. helveticus TS206组丰度更高。在53 个差异OTUs中一共有10 个OTUs属于毛螺菌科,其中有5 个(OTU1 248、OTU801、OTU771、OTU1 355、OTU 1 295)在L. helveticus TS206组富集,另外5 个(OTU468、OTU462、OTU1 238、OTU952、OTU659)在对照组中富集,这些均说明即使属于同一分类水平的物种在肠道中可能会发挥不同的功能。
2.4 与L. helveticus TS206相关的肠道关键物种对各组样本肠道微生物整体结构差异的影响
为了探究通过LEfSe分析寻找到53 个关键OTUs对2 组样本的整体分布情况,基于53 个关键OTUs进行主成分分析,结果如图7所示。

图7 关键OTUs对2 组样本菌群结构分布的影响Fig. 7 Effect of key OTUs on gut microbiota structures
通过主成分分析可以发现,基于53 个关键OTUs的相对丰度进行降维分析,L. helveticus TS206组所有样本聚为一类,对照组所有样本也聚为一类,并且2 组在图7中差异显著(P<0.05),这从另一层面上说明这53 个差异显著OTUs可能是造成L. helveticus TS206组与对照组小鼠肠道菌群结构差异的关键物种。
2.5 关键OTUs在2 组样本间互作网络的特征
为了进一步研究53 个差异OTUs在2 组样本间的互作关系的特征,根据53 个OTUs在2 组样本中的丰度信息计算斯皮尔曼相关性系数,根据各个OTUs间的相关系数大小绘制互作网络图,如图8所示。在对照组富集的和在L. helveticus TS206组富集的OTUs之间互作关系紧密,并且在2 组富集的OTUs之间均呈负相关关系,这也充分说明这些差异OTUs在两组样本的肠道中可能存在竞争抑制的关系。并且,只在对照组富集的属于肠杆菌科的OTUs与L. helveticus TS206组多个OTUs具有负相关关系,最为明显的是与属于梭菌目的OTU41有负相关关系,这也提示该梭菌目的OTUs可能是L. helveticus TS206组中关键的OTUs,并能抵制肠杆菌科物种的增殖作用。另外,只在对照组中富集的属于瘤胃球菌科的OTUs也与L. helveticus TS206组多个OTUs呈现负相关关系;此外,在L. helveticus TS206组富集的丰度较高的拟杆菌属的OTUs也与对照组多个OTUs呈现负相关关系。这些在互作网络图中呈现了丰富连接关系的OTUs节点可能是与L. helveticus TS206组和对照组小鼠肠道微生物结构差异有关的关键OTUs,有待于更深入的探讨。

图8 关键OTUs间互作关系网络图Fig. 8 Interaction network among key OTUs
3 讨 论
人体肠道有种类繁多的微生物,而这些微生物与机体一直处于相互作用的动态平衡中,一旦这种平衡失调就会造成肠道疾病甚至是系统性的慢性疾病。在肠道微生物与宿主相互作用的过程中,宿主自身的基因型、年龄和免疫系统以及饮食结构和营养的摄入等因素共同影响着肠道菌群的形成及多样性组成[18-20]。食品的摄入一方面会改变肠道菌群的结构,另一方面食品中难以被人体吸收的成分通过肠道菌群的作用影响食品的营养。而益生菌作为当前人们日常生活中常用的食品,能够有效地维持机体与肠道菌群处于相互作用的动态平衡中。
本研究发现各组小鼠肠道菌群在门水平上的组成基本无差异,都以拟杆菌门和硬壁菌门为主,相对丰度超过95%,且硬壁菌门丰度最高,超过70%。有其他研究发现,在正常人和小鼠肠道中拟杆菌门和硬壁菌门是两大优势菌门,几乎占肠道所有细菌的90%以上[21-22],并且硬壁菌门是最为优势的一类菌,大多数属于梭菌纲[23],其次是丰度较低的放线菌门和变形菌门[24-25]。在本实验中,L. helveticus TS206组小鼠肠道菌群随着灌胃时间的延长,拟杆菌门丰度有降低的趋势,而硬壁菌门有升高的趋势。
属于硬壁菌门的梭菌目的球形梭菌和柔嫩梭菌亚群中,大多数细菌是产丁酸盐细菌[26],主要通过丁酸激酶或通过丁酰CoA、乙酰CoA转移酶的作用产生丁酸[27]。丁酸是肠上皮细胞能量的主要来源,而且在调节炎症性反应、细胞增殖和凋亡及抗结直肠癌方面也有重要作用[28-29],例如Faecalibacterium prausnitzii就是典型的丁酸产生菌,具有明显的抗炎作用[30]。本研究发现各组小鼠肠道中丰度最高的是梭菌目细菌,通过LEfSe分析发现梭菌目是L. helveticus TS206造成小鼠肠道菌群结构差异的关键菌属,且在L. helveticus TS206组小鼠肠道中丰度更高,这表明L. helveticus TS206可能会通过增加肠道中梭菌目中丁酸产生菌的数量来维持肠道菌群结构的稳定和肠道的健康。另外,也有研究显示梭菌在机体肠道中可以通过分泌短链脂肪酸来诱导和调节调节性T细胞的增殖和分化来塑造肠道内稳定的免疫系统[31-32],在本实验中,L. helveticus TS206诱导的肠道梭菌目细菌的增加可能会对稳定肠道免疫平衡有重要作用。但是由于测序片段较短、分类地位不够明确,有待进一步研究这些梭菌目细菌的具体分类地位。
此外,本实验还发现在L. helveticus TS206灌胃后,丰度显著增加的菌属中大部分是对肠道健康有益的菌属,而在对照组中一些对机体健康不利的细菌丰度较高。例如,由LEfSe分析得知L. helveticus TS206组中跟丁酸产生相关的梭菌目细菌具有更高的丰度,而瘤胃菌科只在对照组中富集。另一方面,在对照组中小鼠肠道中肠杆菌科细菌丰度显著高于L. helveticus TS206组,这个结果也通过传统的平板培养计数实验得到验证。肠杆菌科细菌是常见的内毒素产生菌,能够诱导机体产生炎症性反应,是典型的条件致病菌[21]。王友湘[14]、Frece[15]等在先前的研究中也发现L. helveticus灌胃能够显著增加肠道内乳酸菌和双歧杆菌的数量,抑制肠杆菌的增殖,这与本研究得到的结论相似。
在本研究中,还鉴定出与L. helveticus TS206灌胃显著相关的其他菌属。目前对这些菌属的研究较少,而且对这些菌属的具体作用也不明确。例如,分属于毛螺菌科和瘤胃菌科的OTUs与2 组小鼠肠道菌群差异都显著相关,而且在L. helveticus TS206组小鼠肠道中丰度显著增加的毛螺菌科OTUs数更多。有研究显示,瘤胃菌科细菌在结直肠癌高风险人群肠道内要高于低风险人群[33]。另一项研究也描述了毛螺菌科细菌与结直肠癌的相关性,研究者在评估结直肠癌与肠道菌群的相关性是发现腺瘤患者的一些OTUs相对丰度较高,包括与瘤胃菌科、假单胞菌属和紫单胞菌科相关的OTUs,而与拟杆菌属、毛螺菌科、梭菌目及其他梭菌属相关的OTUs则相对丰度较低[34]。但是,瘤胃菌科和毛螺菌科细菌都是宿主肠道中常见菌属,因此在本研究中2 组小鼠肠道中都有与之相关的OTUs的相对丰度增高,它们在肠道中的具体功能还应进一步通过功能分析来验证。
4 结 论
本研究通过给健康小鼠长期灌胃L. helveticus TS206,利用Ion torrent PGM测序技术和多变量统计学方法分析得知L. helveticus TS206组与对照组小鼠肠道菌群结构具有显著差异。根据测序的数据分析,得到了以下几点提示:从门的水平上分析,各组小鼠肠道粪便中的主要菌群主要由硬壁菌门和拟杆菌门组成,同时L. helveticus TS206能够一定程度上增加肠道微生物中硬壁菌门的相对丰度,并伴随着拟杆菌门相对丰度的降低;从科水平上分析,丰度占优势地位的细菌主要为S24-7、毛螺菌科、瘤胃球菌科、理研菌科和拟杆菌科;此外,结合门水平分析结果,L. helveticus TS206组拟杆菌门细菌丰度的降低是由理研菌科和S24-7科丰度降低引起的;同时,与对照组相比,L. helveticus TS206组肠杆菌科细菌数量具有降低趋势。另外,经LEfSe分析筛选出L. helveticus TS206组与对照组间具有显著差异的53 个关键OTUs,其中肠杆菌科和瘤胃菌科在对照组中富集,梭菌目在L. helveticus TS206组中具有更高的丰度;经主成分分析证实这53 个关键OTUs可能是造成L. helveticus TS206组与对照组小鼠肠道菌群结构差异的关键物种;且通过互作网络图可知,2 组富集的OTUs之间均呈负相关关系,可见这些差异OTUs在2 组样本的肠道中可能存在竞争抑制的关系。因此,研究经生物信息学分析,初步显示L. helveticus TS206可以通过抑制肠道有害微生物的生长、促进有益菌的增殖来维持肠道菌群结构的稳态。然而,研究尚需进一步的定量PCR等验证工作,以充分证实L. helveticus TS206对肠道菌群的调控能力。
[1] LEDERBERG J. Infectious history[J]. Science, 2000, 288: 287-293.DOI:10.1126/science.288.5464.287.
[2] 李旻. 人体肠道菌群结构与宿主代谢的相关性研究[D]. 上海: 上海交通大学, 2009: 15-18.
[3] GOODMAN A L, MCNULTY N P, ZHAO Y, et al. Identifying genetic determinants needed to establish a human gut symbiontin its habitat[J]. Cell Host and Microbe, 2009, 6(3): 279-289. DOI:10.1016/j.chom.2009.08.003.
[4] LEE Y K, MAZMANIAN S K. Has the microbiota played a critical role in the evolution of the adaptive immune system?[J]. Science,2010, 330: 1768-1773. DOI:10.1126/science.1195568.
[5] ZHU Y, MICHELLE T, JOBIN C, et al. Gut microbiota and probiotics in colon tumorigenesis[J]. Cancer Letters, 2011, 309(2): 119-127.DOI:10.1128/mSphere.00001-15.
[6] TURNBAUGH P J, HAMADY M, YATSUNENKO T, et al. A core gut microbiome in obese and lean twins[J]. Nature, 2008, 457: 480-484. DOI:10.1038/nature07540.
[7] BÄCKHED F, MANCHESTER J K, SEMENKOVICH C F, et al.Mechanisms underlying the resistance to diet-induced obesity in germfree mice[J]. Proceedings of the National Academy of Sciences, 2007,104(3): 979-984. DOI:10.1073/pnas.0605374104.
[8] WEN L, LEY R E, VOLCHKOV P Y, et al. Innate immunity and intestinal microbiota in the development of type 1 diabetes[J]. Nature,2008, 455: 1109-1113. DOI:10.1038/nature07336.
[9] QIN J J, LI Y R, CAI Z M, et al. A metagenome-wide association study of gut microbiota in type 2 diabetes[J]. Nature, 2012, 490: 55-60.
[10] VÉTIZOU M, PITT J M, DAILLÈRE R, et al. Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota[J].Science, 2015, 350: 1079-1084. DOI:10.1007/s00109-016-1401-8.
[11] SALMINEN S J, GUEIMONDE M, ISOLAURI E. Probiotics that modify disease risk[J]. Nutrition, 2005, 135(5): 1294-1298.
[12] 张和平. 我国益生乳酸菌研究与产业化现状及发展对策[J]. 生物产业技术, 2009(6): 53-55. DOI:10.3969/j.issn.1674-0319.2009.06.005.
[13] 叶雷, 闫亚丽, 陈庆森, 等. 高通量测序技术在肠道微生物宏基因组学研究中的应用[J]. 中国食品学报, 2016, 16(7): 216-223.DOI:10.16429/j.1009-7848.2016.07.029.
[14] 王友湘, 陈庆森. 瑞士乳杆菌对小鼠肠道微生物区系的影响[J]. 食品科学, 2008, 29(9): 542-546. DOI:10.3321/j.issn:1002-6630.2008.09.129.
[15] FRECE J, KOS B, SVETEC I K, et al. Synbiotic effect of Lactobacillus helveticus M92 and prebiotics on the intestinal microflora and immune system of mice[J]. Dairy Research, 2009,76(1): 98-104. DOI:10.1017/S0022029908003737.
[16] TAVERNITI V, GUGLIELMETTI S. Health-promoting properties of Lactobacillus helveticus[J]. Frontiers in Microbiology, 2012, 3: 392.DOI:10.3389/fmicb.2012.00392.
[17] SINGH N, ARIOLI S, WANG A, et al. Impact of Bifidobacterium bifidum MIMBb75 on mouse intestinal microorganisms[J]. FEMS Microbiology Ecology, 2013, 85(2): 369-375. DOI:10.1111/1574-6941.12124.
[18] LEY R E, LOZUPONE C A, HAMADY M, et al. Worlds within worlds: evolution of the vertebrate gut microbiota[J]. Nature Reviews Microbiology, 2008, 6(10): 776-788. DOI:10.1371/journal.pgen.1001314.
[19] ZHANG C H, ZHANG M H, PANG X Y, et al. Structural resilience of the gut microbiota in adult mice under high-fat dietary perturbations[J].ISME Journal, 2012, 6(10): 1848-1857.
[20] TURNBAUGH P J, BACKHED F, FULTON L, et al. Diet-induced obesity is linked to marked but reversible alterations in the mouse distal gut microbiome[J]. Cell Host and Microbe, 2008, 3(4): 213-223.DOI:10.1016/j.chom.2008.02.015.
[21] ECKBURG P B, BIK E M, BERNSTEIN C N, et al. Diversity of the human intestinal microbial flora[J]. Science, 2005, 308: 1635-1638.DOI:10.1126/science.1110591.
[22] TURNBAUGH P J, LEY R E, MAHOWALD M A, et al. An obesityassociated gut microbiome with increased capacity for energy harvest[J]. Nature, 2006, 444: 1027-1031. DOI:10.1038/nature05414.
[23] HAYASHI H, SAKAMOTO M, BENNO Y. Phylogenetic analysis of the human gut microbiota using 16S rDNA clone libraries and strictly anaerobic culture-based methods[J]. Microbiology and Immunology,2002, 46(8): 535-548.
[24] FEI N, ZHAO L P. An opportunistic pathogen isolated from the gut of an obese human causes obesity in germfree mice[J]. ISME Journal,2013, 7(4): 880-884. DOI:10.1038/ismej.2012.153.
[25] SHKOPOROV A N, KHOKHLOVA E V, KULAGINA E V, et al.Application of several molecular techniques to study numerically predominant Bifidobacterium spp. and Bacteroidales order strains in the feces of healthy children[J]. Bioscience, Biotechnology and Biochemistry, 2008, 72(3): 742-748. DOI:10.1271/bbb.70628.
[26] PRYDE S E, DUNCAN S H, HOLD G L, et al. The microbiology of butyrate formation in the human colon[J]. FEMS Microbiology Letters, 2002, 217(2): 133-139. DOI:10.1016/j.cam.2009.02.103.
[27] FUKUDA S, TOH H, HASE K, et al. Bif i dobacteria can protect from enteropathogenic infection through production of acetate[J]. Nature,2011, 469: 543-547. DOI:10.1038/nature09646.
[28] VANHOUTVIN S A, TROOST F J, HAMER H M, et al. Butyrateinduced transcriptional changes in human colonic mucosa[J]. PLoS ONE, 2009, 4(8): 6759-6766. DOI:10.1371/journal.pone.0006759.
[29] FUNG K Y, COSGROVE L, LOCKETT T, et al. A review of the potential mechanisms for the lowering of colorectal oncogenesis by butyrate[J]. British Journal of Nutrition, 2012, 108(5): 820-831.DOI:10.1017/S0007114512001948.
[30] SOKOL H, PIGNEUR B, WATTERLOT L, et al. Faecalibacterium prausnitzii is an anti-inf l ammatory commensal bacterium identif i ed by gut microbiota analysis of Crohn disease patients[J]. Proceedings of the National Academy of Sciences, 2008, 105(43): 16731-16736.
[31] ATARASHI K, TANOUE T, SHIMA T, et al. Induction of colonic regulatory T cells by indigenous Clostridium species[J]. Science, 2011,331: 337-341. DOI:10.1053/j.gastro.2011.07.013.
[32] FURUSAWA Y, OBATA Y, FUKUDA S, et al. Commensal microbederived butyrate induces the differentiation of colonic regulatory T cells[J]. Nature, 2013, 506: 446-450. DOI:10.1038/nature12721.
[33] MOORE W E, MOORE L H. Intestinal floras of populations that have a high risk of colon cancer[J]. Applied and Environmental Microbiology, 1995, 61(9): 3202-3207.
[34] ZACKULAR J P, ROGERS M A, RUFFIN M T, et al. The human gut microbiome as a screening tool for colorectal cancer[J]. Cancer Prevention Research, 2014, 7(11): 1112-1121. DOI:10.1158/1940-6207.CAPR-14-0129.
Modulation of Probiotic Lactobacillus helveticus on Gut Microbiota in Mice
ZANG Kaili1, JIA Yan1, CUI Wenjing2, MA Xinying2, WANG Yong1, ZHAO Linsen2, ZHAO Pei1, YE Lei1, YAN Yali1,*, CHEN Qingsen1,*
(1. Tianjin Key Laboratory of Food Biotechnology, College of Biotechnology and Food Science, Tianjin University of Commerce,Tianjin 300134, China; 2. Hebei Inatural Biological Technical Company, Shijiazhuang 050899, China)
Objective: To explore the role of the probiotic strain Lactobacillus helveticus TS206 in regulating the intestinal microf l ora in mice and to unravel the mechanisms by which probiotics can maintain intestinal fl ora homeostasis. Methods:Male BALB/c mice were divided into probiotic, control and blank groups. The probiotic and control groups were respectively gavaged with L. helveticus suspension and an equal volume of normal saline once a day for 7 consecutive weeks, respectively. The blank group did not receive gavage. Stool samples were collected every week for extracting genomic DNA by means of a commercial kit. The V6 variable region of the 16S rRNA gene was subjected to highthroughput sequencing by using the Ion torrent personal genome machine system and the obtained data were analyzed using bioinformatic tools and multivariable statistical analysis. Results: All sequence reads were delineated into 1 617 operational taxonomic units (OTUs) at a 97% similarity level, belonging to 8 major bacterial phyla, with Firmicutes and Bacteroidetes being the dominant ones in all groups, accounting for 97.49% of the total number of sequences, amongwhich Firmicutes showed the highest abundance of more than 70%. The strain S24-7, Lachnospiraceae, Ruminococcaceae,Rikenellaceae, and Bacteroidaceae were the dominant bacterial families, and the Enterobacteriaceae population in the probiotic group was lower than in the control group and fell during the administration period. Totally 53 key OTUs were signif i cantly associated with the bacterial community structure in the intestine of the two groups of mice as determined by linear discriminant analysis effect size (LEfSe) analysis, of which, 27 OTUs were enriched in the control group, belonging to Enterobacteriaceae, Ruminococcaceae, Lachnospiraceae and Clostridium, and 26 OTUs in the L. helveticus TS206 group,belonging to Clostridium, whose abundance level was higher than in the control group, Lachnospiraceae and Bacteroides.Principal component analysis showed that the fecal microbial communities of the probiotic group were completely separated from those of the control group. Overall there were signif i cant differences in the structure of intestinal fl ora between the two groups, and the OTUs in the groups showed a negative correlation. Conclusion: L. helveticus TS206 can change the structure of intestinal fl ora, inhibiting the growth of harmful microbes, promoting the proliferation of benef i cial bacteria proliferation and consequently maintaining the structure of the intestinal fl ora at a steady state.
Lactobacillus helveticus TS206; gut microbiota; Ion torrent personal genome machine sequencing technology;bioinformatics; multivariate statistical analysis
10.7506/spkx1002-6630-201801024
TS201.3
A
1002-6630(2018)01-0156-09
臧凯丽, 贾彦, 崔文静, 等. 瑞士乳杆菌调控小鼠肠道菌群变化规律的研究[J]. 食品科学, 2018, 39(1): 156-164.
DOI:10.7506/spkx1002-6630-201801024. http://www.spkx.net.cn
ZANG Kaili, JIA Yan, CUI Wenjing, et al. Modulation of probiotic Lactobacillus helveticus on gut microbiota in mice[J]. Food Science, 2018, 39(1): 156-164. (in Chinese with English abstract)
10.7506/spkx1002-6630-201801024. http://www.spkx.net.cn
2017-04-10
国家自然科学基金面上项目(31071522)
臧凯丽(1993—),女,硕士研究生,研究方向为生物活性物质与肠道健康。E-mail:1623249305@qq.com
*通信作者简介:阎亚丽(1962—)女,副教授,硕士,研究方向为发酵生物技术、微生物学。E-mail:yyali@tjcu.edu.cn
陈庆森(1957—),男,教授,硕士,研究方向为发酵生物技术、食源性生物活性物质与肠道健康。E-mail:chqsen@tjcu.edu.cn
