α-硝基酮类的合成
2017-12-13张寒青潘馨慧张珂王航宇王金辉
张寒青 ,潘馨慧 ,张珂 ,王航宇 ,王金辉 ,2*
(1石河子大学药学院,新疆 石河子832002;2沈阳药科大学中药学院,辽宁 沈阳110016)
α-硝基酮类的合成
张寒青1,潘馨慧1,张珂1,王航宇1,王金辉1,2*
(1石河子大学药学院,新疆 石河子832002;2沈阳药科大学中药学院,辽宁 沈阳110016)
为找到一种简便易行的方法对α-硝基酮类化合物进行合成。采用取代的苯甲酰氯和硝基甲烷为原料,经过两步反应来获得有机反应中重要的中间体α-硝基酮。结果显示,通过该方法合成了12种取代的硝基苯乙酮,其产率在68%-95%之间,结构通过1H NMR验证正确。由此可知,使用取代的苯甲酰氯和硝基甲烷为原料,可以获得相应的硝基酮,并有较为满意的产率。
α-硝基酮;合成;酰氯;硝基甲烷
由于分子结构的特殊性,α-硝基酮作为重要的合成反应中间体[1-6],以它们为原料可制备硝基环己烯、硝酮、螺缩醛、β-硝基醇、ω-硝基醇、β-氨基醇、ω-氨基酸、α-羟基酮等多种合成砌块[7]。
α-硝基酮的合成方法已经较为成熟,从醛、酮、羧酸、烯烃或烷基酯[8-9]出发,均可以得到相应的α-硝基酮化合物。
1955年,C.D.Hurd[10]报道了以醛为原料,但该反应产率不高,仅5%-20%。之后以醛为原料获得硝基酮的反应则存在步骤繁琐[11],或实验条件要求过高[7]的缺点。而已报道的以酮为底物获得硝基酮的反应则对底物要求较为苛刻,环酮为底物[12]或α碳必须有吸电子基的存在时[13]反应才可以发生。而以羧酸[14]为原料,则多需要经长时间的回流才能获得相应的α-硝基酮。而已有的合成硝基苯乙酮的方法,为1990年A.Kunai[15]以苯乙烯为原料所获得,但步骤繁琐。
通过文献的查阅,我们发现寻找合成α-硝基酮的方法仍是一个值得研究的课题。
本文以硝基甲烷和苯甲酰氯(或的苯基酰氯)为原料进行反应,反应全过程不需要经过加热回流等条件,且不需要添加金属催化剂,在温和的条件下,便可进行实验,同时,可以获得满意的产率,产物通过1H NMR表征。
1 材料与方法
1.1 仪器与试剂
仪器:Varian inova-400型和Varian inova-600型核磁共振波谱仪(氘代二甲基亚砜为溶剂)。
试剂:二氯甲烷、二甲基亚砜、三乙胺、乙酸乙酯购自天津市富宇精细化工有限公司,咪唑和硝基甲烷购自成都市科龙化工试剂厂,氢化钠购自上海晶纯生化科技股份有限公司(在矿物油中 含60%),无水硫酸镁购自天津市盛奥化学试剂有限公司,苯甲酰氯购自天津市光复精细化工研究所,2-甲基苯基酰氯,3-甲基苯甲酰氯、3-溴苯甲酰氯、3-氯苯甲酰氯、4-氯苯甲酰氯购自阿达玛斯试剂有限公司,4-甲基苯甲酰氯、4-溴苯甲酰氯、4-甲氧基苯甲酰氯、4-苯基苯甲酰氯购自萨恩化学技术有限公司,4-叔丁基苯甲酰氯、2-溴苯甲酰氯购自阿法埃莎化学有限公司,试剂均为分析纯,无水溶剂为所购试剂通过溶剂除水系统除水。硅胶购自青岛海洋化工厂。
1.2 合成
中间产物b的合成:咪唑(6 mmoL)和三乙胺(6 mmoL)加入到干燥的CH2Cl2,然后再搅拌的溶液中缓慢滴加酰氯(5 mmoL),溶液在室温下反应30 min。反应液过滤,溶液快速用冷水萃取(150 mL×3),有机相用无水硫酸镁干燥,过滤,浓缩,得粗产物N-酰基咪唑,此粗产物可直接用于合成2-硝基苯乙酮,不用纯化。
化合物c的合成:硝基甲烷(5 mmoL)和氢化钠(11 mmoL)加入 DMSO(25 mL),在 10 ℃下反应30 min。将得到的N-酰基咪唑粗产物(5 mmoL)溶于DMSO(25 mL),逐滴的加入反应溶剂中,溶液在10℃下反应30 min,之后在室温下反应12 h。反应液用乙酸乙酯(150 mL)稀释,加入1 N HCl(150 mL),之后用乙酸乙酯萃取(150 mL×3),合并有机相,之后用冷水萃取(150 mL×3),用无水硫酸镁干燥,过滤,浓缩。二氯甲烷/石油醚(v/v=1/1)为洗脱剂过硅胶柱,得相应的2-硝基苯乙酮。
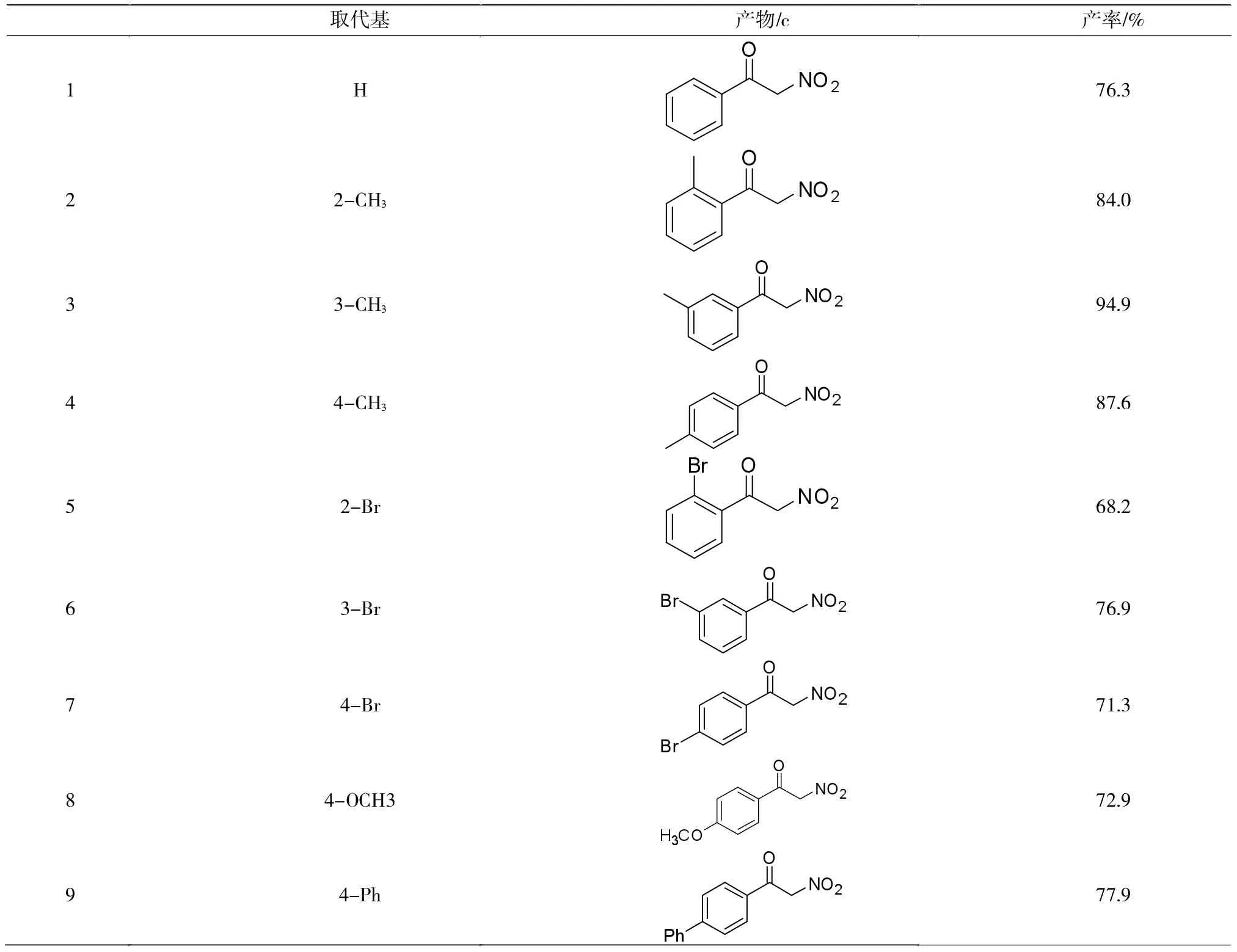
表1 化合物c的合成Tab.1 Synthesis of compound C
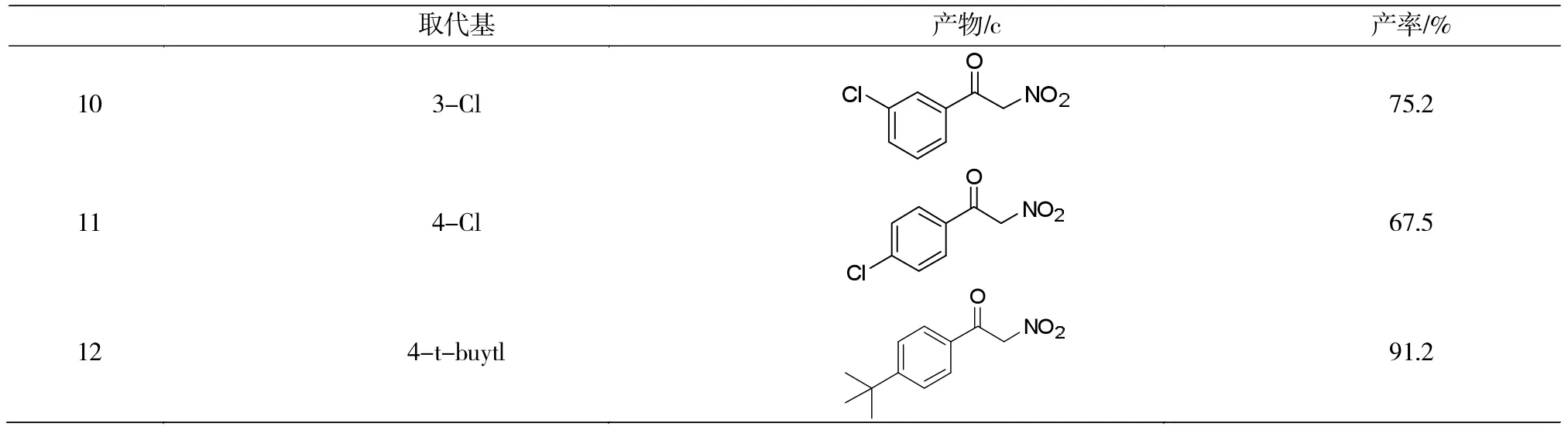
续表1
由表1可以看出,此方法对不同的取代基均有较好的兼容性,反应得到满意的产率。苯环上的取代基对反应有较大的影响,间甲基取代是产率最高,可达到95%,而在邻位存在溴原子和对位存在氯原子时,反应产率仅为68%。所以可知苯环存在供电子基团时,更利于反应的进行,产率有所增加,并且处于间位的供电子基团比处于邻对位的更有利于反应,而吸电子基团的存在较不利于反应进行。当苯环对位有苯环、叔丁基等大位阻基团取代时,反应效率不受影响,且由于叔丁基的供电子效应,产率明显提高,提示此方法对大位阻基团的兼容性较好,有望应用在复杂天然产物的合成中。

图1 α-硝基酮合成路线图Fig.1 The route of synthesis for alpha nitro ketone
1.3 化合物c的谱图分析
2-硝基 -1-苯乙酮(1c)1H NMR(600 MHz,CDCl3) δ 7.91(d,J=7.4 Hz,2H),7.72(t,J=7.5 Hz,1H),7.57(t,J=7.8 Hz,2H),5.92(s,2H)。
2-硝基 (2-甲基)苯乙酮 (2c)1H NMR(400 MHz,DMSO) δ 7.84 (dd,J=8.1,1.3 Hz,1H),7.57(td,J=7.6,1.3 Hz,1H),7.40 (dt,J=7.2,3.4 Hz,2H),6.45(s,2H),2.49(s,3H)。
2-硝基 (3-甲基)苯乙酮 (3c)1H NMR(400 MHz,DMSO) δ 7.87-7.84 (m,1H),7.84-7.82(m,1H),7.42(s,1H),7.40(d,J=0.6Hz,1H),6.50(s,2H),2.41(s,3H)。
2-硝基 (4-甲基)苯乙酮 (4c)1H NMR(400 MHz,DMSO) δ 7.81-7.71 (m,2H),7.57(d,J=7.6 Hz,1H),7.49 (t,J=7.6 Hz,1H),6.52(s,2H),2.40(s,3H)。
2-硝基(2-溴)苯乙酮(5c)1H NMR(400 MHz,DMSO) δ 7.87(dt,J=6.6,3.6 Hz,1H),7.84-7.80(m,1H),7.60-7.52(m,2H),6.43(s,2H)。
2-硝基(3-溴)苯乙酮(6c)1H NMR(400 MHz,DMSO) δ 8.11 (t,J=1.8 Hz,1H),7.95 (dddd,J=8.0,5.0,1.8,1.0Hz,2H),7.57 (t,J=7.9Hz,1H),6.56(s,2H)。
2-硝基(4-溴)苯乙酮(7c)1H NMR(400 MHz,DMSO) δ 7.89-7.85 (m,2H),7.85-7.81(m,2H),6.53(s,2H)。
2-硝基(4-甲氧基)苯乙酮(8c)1H NMR(400 MHz,DMSO) δ 7.94-7.93 (m,1H),7.92-7.90(m,1H),7.15-7.12 (m,1H),7.12-7.10 (m,1H),6.47(s,2H),3.88(s,3H)。
2-硝基 (4-苯基)苯乙酮 (9c)1H NMR(400 MHz,DMSO) δ 8.05-8.01 (m,2H),7.93-7.89(m,2H),7.80-7.77 (m,2H),7.52 (tt,J=6.1,1.7 Hz,2H),7.48-7.43(m,1H),6.58(s,2H)。
2-硝基 (3-氯)苯乙酮 (10c)1H NMR(400 MHz,DMSO) δ 7.98 (t,J=1.9 Hz,1H),7.93-7.88(m,1H),7.85-7.80(m,1H),7.64(t,J=7.9 Hz,1H),6.57(s,2H)。
2-硝基 (4-氯)苯乙酮 (11c)1H NMR(400 MHz,DMSO) δ 7.98-7.95 (m,1H),7.95-7.93(m,1 H),7.71-7.69(m,1H),7.68-7.66(m,1H),6.54(s,2H)。
2-硝基(4-叔丁基)苯乙酮(12c)1H NMR(400 MHz,DMSO) δ 7.92-7.90 (m,1H),7.89-7.87(m,1H),7.63-7.61 (m,1H),7.61-7.59 (m,1H),6.51(s,2H),1.31(s,9H)。
2 结果与讨论
本文以苯甲酰氯和硝基甲烷为原料,两步反应合成硝基酮,该反应条件温和,原料易得,反应简单易操作,具有较高收率。
研究了苯环上取代基的不同对反应产物产率的影响,结果显示,该方法对吸电子基团、供电子基团及大位阻基团均有较好的兼容性,应用广泛。此外,苯环上间位有取代基时,产率要比邻对位有取代基存在时高,同时,供电子基团存在时更利于反应的进行。
[1]Lian Z,Friis S D,Skrydstrup T.Palladium-catalysed carbonylative α-arylation of nitromethane[J].Chem Commun:camb,2015,51(17):3600-3603.
[2]Gangadhara C R,Rajeshwar R G,Ganesh Y S,et al.ChemInform abstract:effect of aqueous polyethylene glycol on 1,3-Dipolar cycloaddition of benzoylnitromethane/Ethyl 2-Nitroacetate with dipolarophiles:green synthesis of Isoxazoles and Isoxazolines.[J].Cheminform,2014,356(1):160-164.
[3]Usami K,Nagasawa Y,Yamaguchi E,et al.Intermolecular cyclopropanation of styrenes using iodine and visible light via carboniodine bond cleavage[J].Organic Letters,2016,18(1):8-11
[4]Baglieri A,Meschisi L,De Sarlo F,et al.Competitive copper catalysis in the condensation of primary nitro compounds with terminal alkynes:synthesis of isoxazoles[J].European Journal of Organic Chemistry,2016,16(27):4643-4655.
[5]Irwan I R,Jiulong S,Chuah G K,et al.Cobalt(II)-Catalyzed electrophilic alkynylation of 1,3-Dicarbonyl compounds to form polysubstituted furans via,π–π activation[J].Advanced Synthesis&Catalysis,2015,357(4):719-726.
[6]Berestovitskaya V M,Baichurin R I,Aboskalova N I,et al.Geminally activated nitroethenes in reactions with sodium azide.synthesis of functionalized 1,2,3-triazoles[J].Russian Journal of General Chemistry,2016,86(6):1266-1273.
[7]赵昌燕,张向阳,徐中轩,等.α-硝基酮的合成[J].合成化学,2009,17(1):125-127.Zhao C Y,Zhang X Y,Xu Z X,et al.Synthesis of alpha nitro ketone[J].Synthesis Chemistry,2009,17(1):125-127.
[8]Shahi S P,Gupta A,Pitre S V,et al.ChemInform abstract:chemistry of nitro compounds.part 9.trimethylsilylnitratechromium trioxide and trimethylsilylnitrate-DMSO:novel reagent systems for one step conversion of olefins into α-Nitro ketones and cyclic ethers into lactones[J].Cheminform,1999(64):4509-4511.
[9]Field G F,Zally W J.Umsetzungen von dilithio-nitroalkanen und-allylnitroderivaten mit carbonylverbindungen[J].Synthesis,1979(4):295-296.
[10]Hurd C D,Nilson M E.Aliphatic Nitro Ketones[J].Journal of Organic Chemistry,1955(20):927–936.
[11]Melot J M,Texier-Boullet F,Foucaud A.ChemInform abstract:preparation and oxidation of α-nitro alcohols with supported reagents[J].Tetrahedron Letters,1986,27(4):493-496.
[12]Dampawan P,Nitro Ketones,Nitration of enol acetates with trifluoroacetic anhydride and ammonium nitrate[J].Synthesis,1983(7):545-546.
[13]Kislyi V P,Laikhter A L,Ugrak B I,et al.ChemInform abstract:synthesis of α-Functional nitro compounds by nitration of activated carbonyl derivatives in a two-phase system.[J].Izvestiya Akademii Nauk,Seriya Khimicheskaya,1994(1):76-79.
[14]Baker D C,Putt S R.ChemInform abstract:C-acylation of nitromethane.a synthetic route to α-Nitroketones[J].Chemischer Informationsdienst,1978,9(40):478-479.
[15]Kunai A,Doi T,Kishimoto T,et al.ChemInform abstract:conversion of styrene to 2,5-Diphenyl-1,4-pyrazine and Related Compounds[J].Cheminform,1990(5):245-248.
Synthesis of α-nitroKetone
Zhang Hanqing1,Pan Xinghui1,Zhang Ke1,Wang Hangyu1,Wang Jinhui1,2*
(1 School of Pharmacy,Shihezi University,Shihezi,Xinjiang 832002,China;2 School of Traditional Chinese Materia Medica,Shenyang Pharmaceutical University,Shenyang, Liaonin 110016,China)
To find a simple and convenient method for the α-nitroKetone synthesis.The substituted benzoyl chloride and nitromethane were used as raw materials in the study;the two step reaction was used to obtain the important intermediate.The results showed that 12 kinds of substituted acetophenone were synthesized by this method,the yield was 68%-95%,and the structure was verified by1H NMR.The conclusion is that by using substituted benzoyl chloride and nitromethane as raw materials,the corresponding nitro group can be obtained,and the yield is satisfactory.
α-nitro ketone;synthesis;chloride;nitromethane
R914.5
A
10.13880/j.cnki.65-1174/n.2017.05.013
1007-7383(2017)05-0602-04
2017-01-19
新疆特种植物药资源教育部重点实验室开放课题(20150203)
张寒青(1991-),女,硕士研究生,专业方向为药物化学。
*通信作者:王金辉(1972-),男,教授,从事中药及天然药物中有效成分的研究,e-mail:15999290001@163.com。
