液相色谱-串联质谱法测定尿液中7种有机磷酸酯代谢物
2017-11-06曾祥英崔君涛赵灵娟于志强
李 佩 曾祥英 崔君涛 赵灵娟 于志强*
1(中国科学院广州地球化学研究所, 有机地球化学国家重点实验室,广东省环境资源利用与保护重点实验室, 广州 510640)2(中国科学院大学, 北京 100049)
液相色谱-串联质谱法测定尿液中7种有机磷酸酯代谢物
李 佩1,2曾祥英1崔君涛1,2赵灵娟1于志强*1
1(中国科学院广州地球化学研究所, 有机地球化学国家重点实验室,广东省环境资源利用与保护重点实验室, 广州 510640)2(中国科学院大学, 北京 100049)
利用液相色谱-三重四极杆质谱(LC-MS/MS)联用技术,建立了人尿液中7种有机磷酸酯代谢产物-有机磷酸二酯的检测方法。针对不同理化性质的有机磷酸二酯,采用固相萃取技术进行富集净化,筛选出高效的固相萃取柱,并对其洗脱条件进行优化; 同时,对流动相和质谱参数进行优化,获取用于各代谢产物定性与定量分析的特征离子对。研究结果表明,前处理采用Oasis WAX固相萃取柱富集、2 mL含5%氨水的甲醇和2 mL甲醇洗脱目标物,除磷酸二乙酯(DEP,17.8%~36.2%)外,其余目标化合物回收率均在60.5%~104.0%之间。在优化的液相色谱条件下,7种化合物可达到完全基线分离。7种有机磷酸二酯的检测限和定量限分别在0.005~0.2 μg/L和0.02~0.5 μg/L之间,日内和日间精密度结果(RSD≤15.4%)表明,本方法具有较好的稳定性和重现性。本方法用于普通人群尿液中有机磷酸二酯的检测,7种化合物在尿液中均有检出,总浓度在0.5~6.7 μg/L之间。
液相色谱-串联质谱; 有机磷酸酯; 尿液; 代谢产物
2017-06-21收稿;2017-07-19接收
本文系国家杰出青年科学基金(No. 41225013)和中国科学院战略性先导科技专项(B类) (No. XDB14010202)资助
* E-mail: zhiqiang@gig.ac.cn
1 引 言
随着溴代阻燃剂的逐步禁用,有机磷酸酯(Organophosphate esters, OPs)作为替代品,近年来其生产和使用量逐步增大[1]。有机磷酸酯作为阻燃剂、增塑剂以及抗发泡剂广泛应用于各行业,如建材、纺织、化工、电子以及石油等行业[2]。研究显示,OPs已成为全球性的环境污染物,在空气、水、灰尘、土壤、沉积物以及生物体中广泛分布[3]。毒理研究证实,OPs具有神经毒性、致癌、致畸、内分泌干扰效应等[4,5],对人体健康具有潜在的严重危害,因此,亟需开展人体OPs的内暴露水平研究,为OPs的健康风险评价提供基础科学数据。
动物实验及体外细胞实验均表明,OPs脱侧链形成磷酸二酯化合物是其在生物体内的主要代谢产物[6,7],OPs通过人体代谢之后,主要通过尿液排出体外。因此,测定人体尿液中OPs的二酯代谢物水平,是目前评价OPs人体内暴露水平的主要方法。总体来看,OPs的人体内暴露研究相对较少,目前主要集中在分析方法的建立和完善上。文献报道的OPs二酯代谢物检测方法主要有两类,一类是目标物衍生化后通过气相色谱-三重四极杆质谱(GC-MS/MS)进行测定,如Schindler等[8,9]利用固相萃取(SPE)对尿液进行初步富集净化后,通过与五氟苄基溴的反应进行衍生化,衍生化产物再次进行SPE净化,之后通过GC-MS/MS检测,该方法前处理较为复杂,不适合于大批量样品的检测。另一类方法是通过液相色谱-三重四极杆质谱(LC-MS/MS)进行测定。Su等[10]建立了超高效液相色谱-三重四极杆质谱检测方法,使用电喷雾负离子源模式进行非卤代磷酸二酯的测定,然后通过重氮甲烷进行衍生化,用电喷雾正离子源模式进行卤代磷酸二酯的测定,显著提高了OPs代谢物的灵敏度。但由于尿液基质复杂,衍生化反应需要较高的技术水平才能确保其稳定性。Cequier等[11]建立了超高效液相色谱-飞行时间质谱仪的直接进样技术,尿液无需处理,3 min内即可完成分析,但基质干扰严重,检测限较高。文献[12,13]建立了SPE-LC-MS/MS检测的方法,同时分析尿液中6种磷酸二酯,尿样经过混合阴离子交换固相萃取柱富集后,可直接进仪器分析,减少了样品前处理步骤。上述方法基本能满足检测需求,但仍存在不足,如基质干扰大,造成样品中检出率低、检测的代谢产物种类有限等。本研究基于对中国环境介质中OPs污染特征的研究结果[14,15],以尿液中7种OPs化合物的二酯代谢物为目标物,拟通过SPE小柱的筛选和优化,确定前处理富集净化方法; 同时进一步优化质谱参数,建立快速简捷的分离富集和定量分析方法。
2 实验部分
2.1仪器与试剂
1100型液相色谱仪(LC,美国Agilent公司); API4000三重四极杆质谱仪(MS/MS,美国AB SCIEX公司); HeraeusTMLabofugeTM200台式离心机(美国Thermo Fisher公司); 12孔固相萃取装置(美国Supeclo公司); Milli-Q Unique-R10超纯水系统(美国Millipore公司); SPE小柱Oasis WAX Extraction Cartridges (60 mg, 3 mL, 美国Waters公司)。
标准物质:磷酸二乙酯(DEP)和DBP(美国ChemSevices公司); 磷酸二苯酯(DPhP)、磷酸二(2-氯乙基)酯(BCEP)、磷酸二(2-氯丙基)酯(BCPP)、回收率指示物D10-DPhP、D8-BCEP和D12-酯BCPP(加拿大Toronto Research Chemicals公司); 磷酸二(1,3-二氯异丙基)(BDCPP, 加拿大Wellington公司); 磷酸二苄基酯(DTP,日本Tokyo Chemical Industry公司)。
甲醇(LC级,德国Merck公司); 乙酸(LC级,美国Tedia公司); 氨水(28%~30%)和无水乙酸钠均购自上海安谱科技公司。实验用水为超纯水。
2.2尿液样品采集
尿液样品委托南方医院于2014年10月至2015年7月间收集,所有志愿者均自愿参加并签署知情同意书,本研究随机挑选10例样本进行检测,对方法进行验证。尿液样本用预先处理过的聚乙烯塑料瓶收集并置于80℃保存。
随机采取实验室志愿者尿液50例,每例取10 mL进行混合,形成混合尿液基质。采用混合尿液基质加标样品对方法进行优化。
2.3样品处理
样品置于室温下解冻后,3000 r/min离心10 min后取上清液; 准确量取2 mL,加入10 ng回收率指示物(D10-DPhP、D8-BCEP和D12-BCPP),加200 μL 0.1 mol/L醋酸钠-醋酸(NaAc-HAc)缓冲溶液(pH 4.5)调节样品至pH 5.0,充分混匀并平衡6~8 h后,过固相萃取柱净化。固相萃取柱依次用2 mL甲醇、2 mL含5%氨水的甲醇和3 mL 0.1 mol/L NaAc-HAc缓冲溶液(pH=4.5)进行活化,上样后,用2 mL 30%甲醇溶液(pH 5.0)淋洗,之后固相萃取柱用氮气吹干,最后用2 mL含5%氨水的甲醇和2 mL甲醇洗脱目标化合物。收集的组分氮吹近干,用50%甲醇溶液溶解并定容至200 μL,进仪器分析。
2.4仪器分析
采用Zorbax SB-C18柱(250 mm × 4.6 mm, 5 μm,美国Agilent公司)为色谱分析柱,甲醇、乙腈和10 mmol 甲酸铵-氨水缓冲溶液(pH 9.2)为流动相,流速0.4 mL/min,柱温32℃,梯度淋洗程序见表1。质谱检测采用电喷雾离子源(ESI),负离子模式,电压4500 V,离子源温度450℃,雾化气压力50 psi,辅助气压力60 psi,窗帘气压力30 psi; 采用多反应监测模式(MRM)。各化合物的保留时间和定性定量离子对见表2。
表1 梯度淋洗程序
Table 1 Gradient elution program of mobile phase
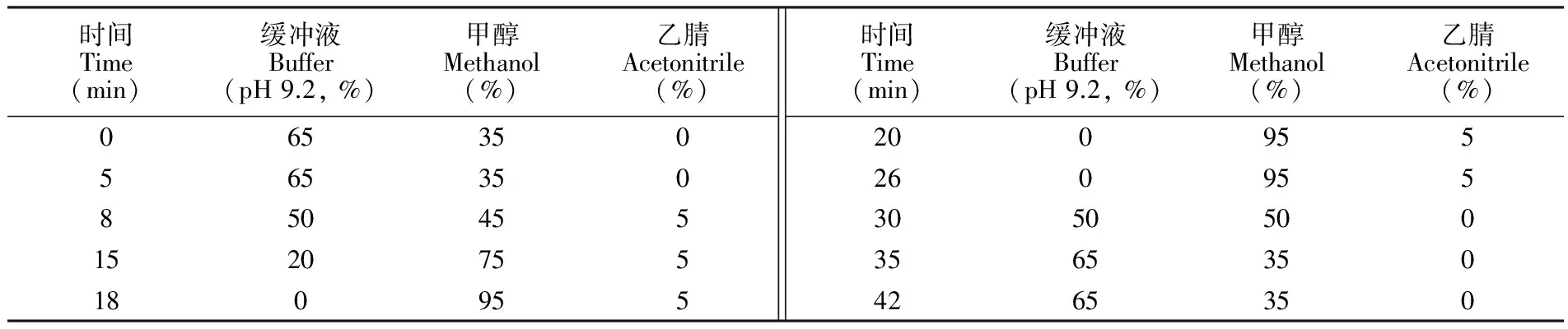
时间Time(min)缓冲液Buffer(pH9.2,%)甲醇Methanol(%)乙腈Acetonitrile(%)0653505653508504551520755180955时间Time(min)缓冲液Buffer(pH9.2,%)甲醇Methanol(%)乙腈Acetonitrile(%)200955260955305050035653504265350
表2 有机磷酸酯代谢物的色谱及质谱参数
Table 2 Instrumental parameters for seven metabolites of OPs and internal standards
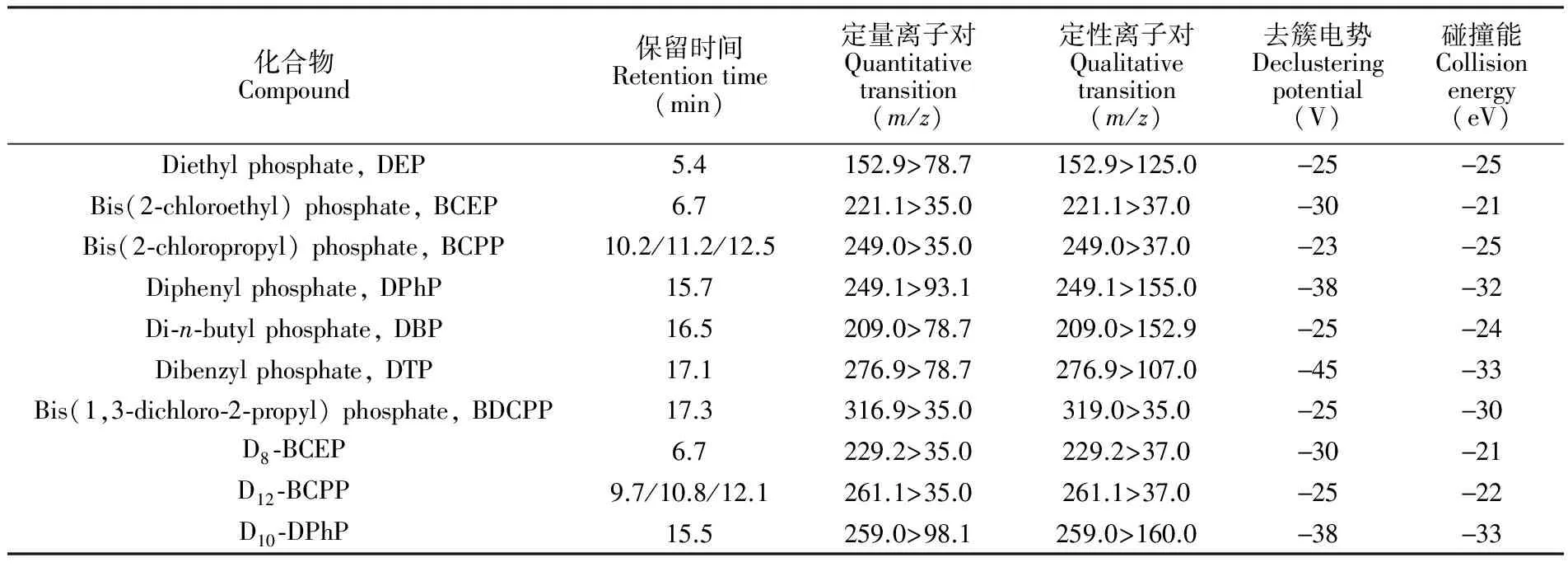
化合物Compound保留时间Retentiontime(min)定量离子对Quantitativetransition(m/z)定性离子对Qualitativetransition(m/z)去簇电势Declusteringpotential(V)碰撞能Collisionenergy(eV)Diethylphosphate,DEP5.4152.9>78.7152.9>125.0-25-25Bis(2⁃chloroethyl)phosphate,BCEP6.7221.1>35.0221.1>37.0-30-21Bis(2⁃chloropropyl)phosphate,BCPP10.2/11.2/12.5249.0>35.0249.0>37.0-23-25Diphenylphosphate,DPhP15.7249.1>93.1249.1>155.0-38-32Di⁃n⁃butylphosphate,DBP16.5209.0>78.7209.0>152.9-25-24Dibenzylphosphate,DTP17.1276.9>78.7276.9>107.0-45-33Bis(1,3⁃dichloro⁃2⁃propyl)phosphate,BDCPP17.3316.9>35.0319.0>35.0-25-30D8⁃BCEP6.7229.2>35.0229.2>37.0-30-21D12⁃BCPP9.7/10.8/12.1261.1>35.0261.1>37.0-25-22D10⁃DPhP15.5259.0>98.1259.0>160.0-38-33
3 结果与讨论
3.1SPE条件优化
固相萃取柱是决定样品富集净化效果的重要因素。本研究选取Oasis WAX、Oasis HLB和Bond Elut C183种小柱进行对照。研究表明,Bond Elut C18柱对磷酸二酯的富集效果不佳,所有考察的目标污染物回收率均小于50%,这可能与磷酸二酯的极性较大、pKa值小相关。通常情况下,磷酸二酯在尿液中以负离子形式存在[11],因此在反相非极性C18柱上的富集效果相对较差; 而Oasis HLB对水溶性较大的DEP和BCEP的回收率分别为19.4%和32.2%,因此选择Oasis WAX弱阴离子柱为固相萃取柱。尽管Oasis WAX有较高的DBP背景污染,但其富集效率和净化效果相对其它小柱更好,可通过扣除背景值的方法进行校正。此结果与van den Eede等研究结果一致,他们也发现Oasis WAX含有一定的DBP背景[12]。Cooper等[13]使用Strata-X-AW柱富集尿液中的磷酸二酯,回收率相对比较稳定; van den Eede等[12]在研究中进一步发现,Strata-X-AW小柱易引发严重的基质干扰; 而Bond Elut NH3柱无法有效富集有机磷酸二酯,在SPE的上样阶段即有80%的目标物流出损失。因此,最终确定使用Oasis WAX柱作为固相萃取柱。

图1 尿液pH值对回收率的影响Fig.1 Effect of urine pH value on recovery
进一步考察了pH值、淋洗溶剂和洗脱溶剂对回收率的影响。由于磷酸二酯的极性较大,且pKa值低(<3.0)[13],正常尿液基质中(pH=5~8),它们均以负离子形式存在,为确保富集效果,固相萃取前需要对尿液pH值进行调节。Oasis WAX是弱阴离子交换柱,被富集的样品溶液比较理想的pH值是比目标物的pKa值大2个pH单位,与比柱吸附剂的pKa(Oasis WAX柱的pKa≈ 6.5)值小2个pH单位之间; 另一方面,使用缓冲溶液稀释样品,降低样品的离子强度,上样后目标物与Oasis WAX柱吸附剂的弱阴离子进行交换而被富集。鉴于此,本研究利用0.1 mol/L NaAc-HAc缓冲溶液(pH=4.5)进行稀释,并对尿液pH值进行优化。如图1所示,当pH=5.0时各目标化合物均有较理想的回收率,因此最终选择用200 μL NaAc-HAc缓冲溶液(pH=4.5)对尿液进行稀释。
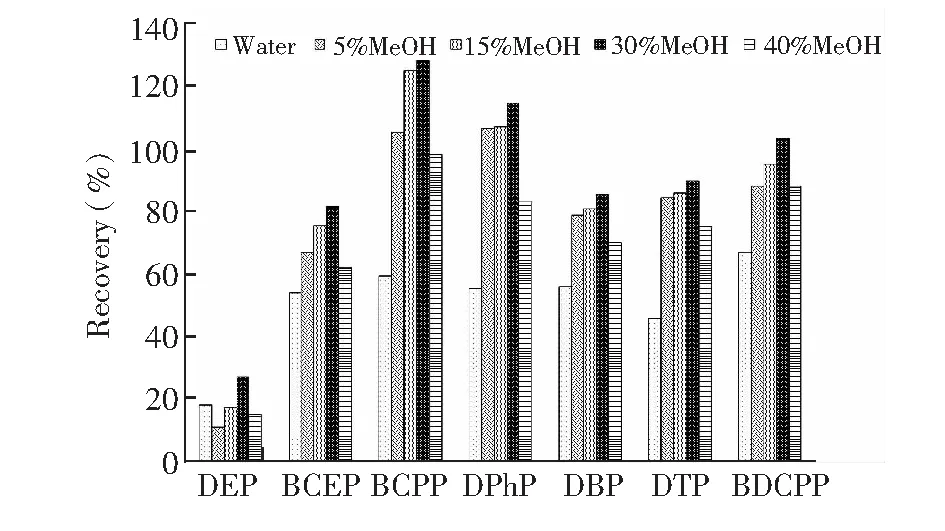
图2 淋洗溶剂对回收率的影响Fig.2 Effect of washing solvent on recovery
在洗脱目标化合物之前的有效淋洗非常重要,可以有效去除杂质,降低生物基质的干扰。图2显示了不同淋洗液对磷酸二酯回收率的影响,各淋洗液均用0.1 mol/L NaAc-HAc缓冲溶液调节pH=5。当洗脱溶剂中甲醇含量在5%~15%时,尿液样品中的杂质无法较好地去除,基质干扰效应相对较强,影响目标化合物的准确测定。当增加淋洗溶液中甲醇的比例增至40%时,目标物在淋洗时流失,造成富集效果显著下降。综合考虑,30% MeOH(pH=5)溶液淋洗,在保证各化合物回收率的条件下,能最大限度洗脱样品中的基质。
弱阴离子交换柱在洗脱时,主要通过中和小柱吸附剂上的电荷对目标物进行洗脱,因此常用高离子强度的缓冲液或带有少量酸或者碱的有机溶剂洗脱,本研究使用甲醇溶液洗脱目标物,且在甲醇中加5% NH3·H2O(pKa= 9.26),用于中和柱内吸附剂上的电荷,削弱目标物与柱吸附剂的离子交换。因此使用2 mL 含5% NH3·H2O的MeOH溶液作为洗脱剂,这与文献[12]中使用的洗脱溶剂一致。本研究表明,在2 mL含5% NH3·H2O的MeOH溶液洗脱之后,再增加2 mL MeOH洗脱,可有效提高DTP的回收率,这可能与DTP的亲脂性较强有关。因此本研究采用2 mL 含5% NH3·H2O的MeOH和2 mL MeOH作为洗脱液。
3.2色谱-质谱条件优化
研究表明[11,12],在流动相水中加入10 mmol/L甲酸铵-氨水缓冲溶液,并调节至pH=9.2,可提高有机磷酸二酯化合物的灵敏度。由于乙腈的洗脱能力较甲醇强,因此在梯度淋洗中加入适量的乙腈可缩短化合物在色谱柱的保留时间,利于污染物实现基线分离,从而避免共溢出造成化合物在离子源处的竞争离子化。
对于各磷酸二酯化合物的质谱参数优化,依据下面的原则:首先,在全扫描模式下(Full scan,m/z=50~500),确定每个目标化合物的特征离子峰; 然后,选择质荷比大(一般为分子离子峰)且灵敏度高的特征碎片离子作为母离子,进行子离子扫描,确定每个目标化合物的定性和定量离子对; 最后,通过多级反应监测模式(MRM),对各化合物定性定量离子对及碰撞能等参数进行优化,最终优化的结果见表2。标准溶液及尿液加标样品中各化合物的总离子流图见图3。从图3可见,本研究所有的目标化合物均达到基线分离。

图3 标准溶液和尿液基质加标样品中OPs代谢物的总离子流图(TIC),其中A为标准溶液中7种OPs代谢物(实线)和3个内标(虚线)的TIC图; B为A图中6~14 min的放大图; C为尿液基质加标样品中OPs化合物的TIC图; D为C图中6~14 min的放大图。Fig.3 Total ion chromatograms for metabolites of OPs. (A) All analytes in standard solution, (B) BCEP and BCPP and their corresponding internal compounds in standard solution, (C) All analytes in spiked urine sample and (D) BCEP and BCPP and their corresponding internal compounds in spiked urine sample
3.3方法评价
从回收率、重复性、线性范围、检出限及基质效应几个方面对建立的方法进行评价。在人体尿液混合基质中分别添加最终浓度为10、 50和100 μg/L的混合标准溶液,结果见表3。除DEP外,其它化合物的回收率均在60.5%~104.0%之间,这可能由于DEP的极性和水溶性较大,在淋洗基质时,很容易随着基质流出,且在色谱分离时,保留时间靠前,受非保留基质的影响较大造成的。DEP的回收率与文献[16]报道的研究结果相近。文献[17]使用乙腈液液萃取尿液中OPs代谢物,其中DEP的平均回收率可达114%,因此液液萃取可能是提高DEP回收率的有效方法。
表3 7种有机磷酸酯代谢物的加标回收率、检测限、定量限及重现性
Table 3 Recovery, limit of detection, limit of quantification and repeatability for seven metabolites of OPs (n=6)

化合物Compound回收率Recovery(%)加标水平Spikinglevel10μg/L50μg/L100μg/L日内精密度Intra⁃dayprecision(%)日间精密度Inter⁃dayprecision(%)检出限LOD(μg/L)定量限LOQ(μg/L)DEP36.2±2.027.8±0.717.8±1.55.711.60.0300.100BCEP63.0±10.460.5±13.574.7±6.59.415.40.1000.300BCPP72.7±14.683.7±12.089.5±9.010.710.70.2000.500DPhP96.3±11.392.3±4.397.1±3.92.010.50.0050.020DBP104.0±1.799.8±8.997.6±2.71.99.50.0060.020DTP91.7±10.898.2±6.193.8±7.91.911.60.0070.020BDCPP91.7±12.283.3±3.279.7±8.34.812.70.0500.200
各化合物在0.5~100 μg/L的浓度范围内呈现良好的线性关系,相关系数(R2)在0.996~0.999之间; 取3倍信噪比(S/N)为检出限,10倍S/N为定量限,各目标物的检出限在0.005~0.200 μg/L之间,定量限在0.020~0.500 μg/L之间。
研究还发现,即使在优化后的分离富集条件以及仪器分析条件下,目标化合物也存在显著的基质效应。通过混合尿液基质加标与溶剂标样同时检测,基质效应计算公式如下:
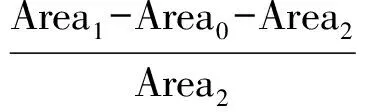
其中, ME为基质效应; Area0为混合基质中目标物检测的响应峰面积; Area1为混合基质加标后目标物检测的响应峰面积; Area2为溶剂标样中目标物检测的响应峰面积。
ME<0, 表示有基质抑制效应; ME>0, 表示有基质促进效应; ME=0, 表示无基质效应。结果表明, 各化合物的基质效应在56%~26%之间,大部分化合均呈现基质抑制效应,其中DEP(56%)和BCEP(23%)受基质抑制效应干扰最大。本研究报道的基质效应显著低于文献[12]的报道值(40%~166%),这可能与本研究所建立的SPE富集净化方法和液相分离相关,高效的前处理方法以及液相分离均可有效去除基质,使得目标污染物得到有效分析。
本研究与文献报道的其它方法比较(表4),不需要进行衍生化,操作简单且具有较低的检出限。本研究首次报道了尿液中DTP的检测。在优化条件下,本研究中各化合物的回收率除DEP较低外(17.8%~36.2%),其它化合物的回收率在60.5%~104.0%之间,日间重复实验结果(RSD ≤15.4%)表明本方法稳定可靠。
3.4实际样品分析
将方法应用于10例人体尿液样品中7种OPs代谢物的分析。尿液中7种OPs代谢物的浓度如表5所示。各化合物在样品中均有检出,除BCPP仅在1例样品中有检出外,其它化合物的检出率均大
表4 本研究方法与文献已报道的方法比较
Table 4 Comparison of method parameters between this method and literature methods for OPs analysis

预处理Samplepreparationa仪器分析Instrumentalanalysis目标物Analyte检测限LOD(μg/L)参考文献Ref.SPE(IsoluteENV+andBondElutPSA+Florisil)GC⁃MS/MSDBP,BCPP,BCEP,DPhP,DmCP,DpCP0.1~1[8,9]SPE(StrataX⁃AW)LC⁃MS/MSBDCPP,DPhP0.008~0.2[13]LLELC⁃MS/MSDEP,DiBP,DnBP,DPhP,DBEP,DEHP0.3~11[17]SPE(OasisWAX)LC⁃MS/MSBCEP,BCPP,BDCPP,DBP,DPhP,BBOEP1.3~25[12]DirectinjectionLC⁃TOFMSBCEP,BCPP,DBP,DPhP,BDCPP,BBOEP0.1~25[11]SPE(ISOLUTEaminopropyl)UPLC⁃MS/MSDBP,DPhP,BBOEP,DEHP,BCEP,BCPP,BDCPP0.02~0.2[10]SPE(OasisWAX)UPLC⁃MS/MSBDCPP,DPhP,BBOEP,BCPP,BCEP,DpCP0.08~0.25[18]SPE(OasisWAX)LC⁃MS/MSDEP,BCEP,BCPP,DBP,DPhP,BDCPP,DTP0.005~0.2本研究Thisworka括号内为固相萃取柱类型。DmCP:di⁃m⁃cresylphosphate;DpCP:di⁃p⁃cresylphosphate;BBOEP:bis(2⁃butoxyethyl)phosphate;DEHP:di(2⁃ethylhexyl)phosphate
表5 10例普通人群尿样中OPs代谢物的体积浓度值(μg/L)
Table 5 Concentrations of metabolites of OPs in 10 samples(μg/L)
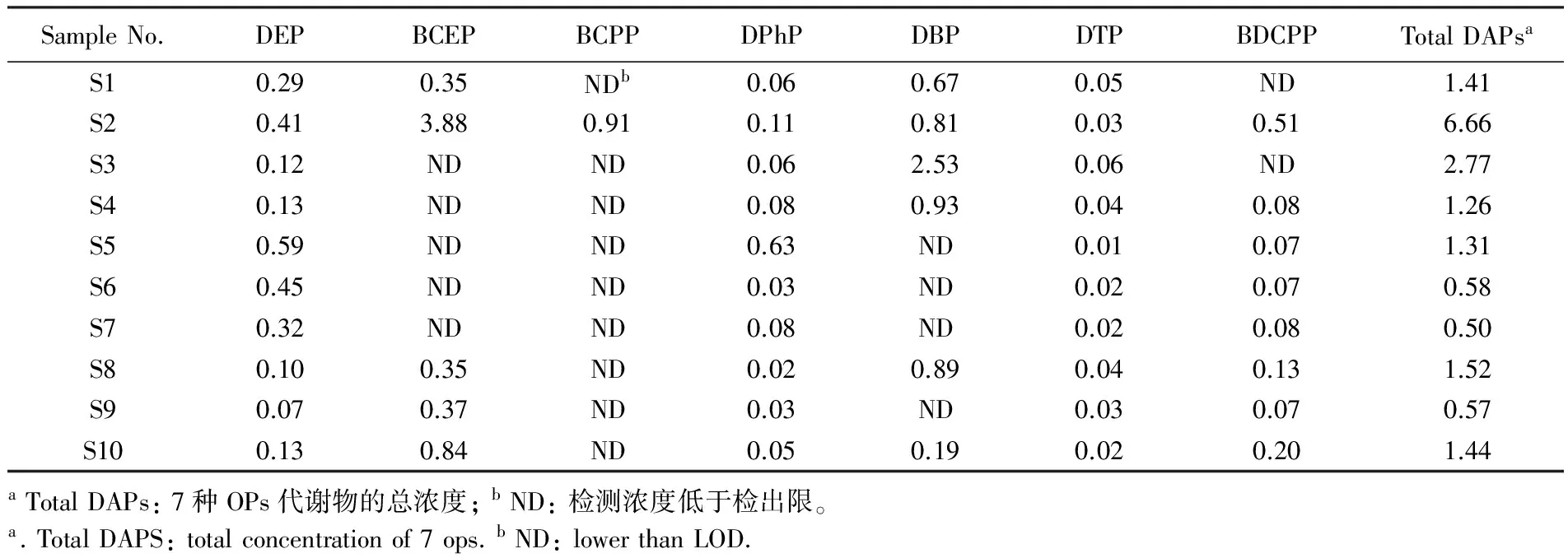
SampleNo.DEPBCEPBCPPDPhPDBPDTPBDCPPTotalDAPsaS10.290.35NDb0.060.670.05ND1.41S20.413.880.910.110.810.030.516.66S30.12NDND0.062.530.06ND2.77S40.13NDND0.080.930.040.081.26S50.59NDND0.63ND0.010.071.31S60.45NDND0.03ND0.020.070.58S70.32NDND0.08ND0.020.080.50S80.100.35ND0.020.890.040.131.52S90.070.37ND0.03ND0.030.070.57S100.130.84ND0.050.190.020.201.44aTotalDAPs:7种OPs代谢物的总浓度;bND:检测浓度低于检出限。a.TotalDAPS:totalconcentrationof7ops.bND:lowerthanLOD.
于等于50%,7种OPs代谢物的总和浓度范围为0.5~6.7 μg/L,其中DEP、BCEP和DBP是检出的主要化合物。OPs的人体内暴露研究可为OPs的健康风险评价提供重要的科学数据。
4 结 论
本研究建立了液相色谱-三重四极杆串联质谱检测人体尿液中7种OPs代谢物的分析方法,前处理采用固相萃取富集净化,2 mL尿液加200 μL 0.1 mol/L醋酸钠-醋酸缓冲溶液稀释,稀释后的样品通过固相萃取柱富集,目标物用2 mL含5%氨水的甲醇和2 mL甲醇洗脱。本方法具有较高的回收率和重现性,可用于大样本量尿液中的OPs代谢物的检测。本研究使用柱容量较大的分析柱对目标物进行色谱分离,减少了基质干扰,提高了卤代OPs代谢物的检测灵敏度。但是相比较非卤代OPs代谢物,卤代OPs代谢物的检测灵敏度偏低,未来需要寻求提高卤代OPs代谢物检测灵敏度的简便方法。
1 Stapleton H M, Klosterhaus S, Eagle S, Fuh J, Meeker J D, Blum A, Webster T F.Environ.Sci.Technol.,2009, 43(19): 7490-7495
2 Marklund A, Andersson B, Haglund P.Chemosphere.,2003, 53(9): 1137-1146
3 van der Veen I, de Boer J.Chemosphere,2012, 88(10): 1119-1153
4 Liu C, Su G, Giesy J P, Letcher R J, Li G, Agrawal I, Li J, Yu L, Wang J, Gong Z.Sci.Rep.,2016, 6: 19045
5 Wei G L, Li D Q, Zhuo M N, Liao Y S, Xie Z Y, Guo T L, Li J J, Zhang S Y, Liang Z Q.EnvironPollut.,2015, 196: 29-46
6 Van den Eede N, Tomy G, Tao F, Halldorson T,Harrad S, Neels H, Covaci A.Chemosphere,2016, 144: 1299-1305
7 Hou R, Xu Y P, Wang Z J.Chemosphere,2016, 153: 78-90
8 Schindler B K, Foerster K, Angerer J.J.Chromatogr.B,2009, 877(4): 375-381
9 Schindler B K, Foerster K, Angerer J.AnalBioanalChem.,2009, 395(4): 1167-1171
10 Su G, Letcher R J, Yu H.J.Chromatogr.A,2015, 1426: 154-160
11 Cequier E, Marce R M, Becher G, Thomsen C.Anal.Chim.Acta,2014, 845: 98-104
12 van den Eede N, Neels H, Jorens P G, Covaci A.J.Chromatogr.A,2013, 1303: 48-53
13 Cooper E M, Covaci A, van Nuijs A L N, Webster T F, Stapleton H M.Anal.Bioanal.Chem.,2011, 401(7): 2123-2132
14 Luo P, Bao L J, Guo Y, Li S M, Zeng EY.J.HazardousMater.,2016, 301: 504-511
15 Zeng X Y, He L X, Cao S X, Ma S T, Yu Z Q, Gui H Y, Sheng G Y, Fu J M.Environ.Toxicol.Chem.,2014, 33: 1720-1725
16 BickerW, Lämmerhofer M, Lindner W.J.Chromatogr.B,2005, 822(1-2): 160-169
17 Reemtsma T, Lingott J, Roegler S.Sci.TotalEnviron.,2011, 409(10): 1990-1993
18 Kosarac I, Kubwabo C, Foster W G.J.Chromatogr.B,2016, 1014: 24-30
This work was supported by the National Natural Science Foundation of China (No. 41225013).
DeterminationofSevenUrinaryMetabolitesofOrganophosphateEstersUsingLiquidChromatography-TandemMassSpectrometry
LI Pei1,2, ZENG Xiang-Ying1, CUI Jun-Tao1,2, ZHAO Ling-Juan1, YU Zhi-Qiang*1
1(StateKeyLaboratoryofOrganicGeochemistry,GuangdongKeyLaboratoryofEnvironmentandResources,GuangzhouInstituteofGeochemistry,ChineseAcademyofSciences,Guangzhou510640,China)2(UniversityofChineseAcademyofSciences,Beijing100049,China)
A simple method was developed for simultaneous determination of seven urinary metabolites of organophosphate esters by liquid chromatography-tandem mass spectrometry (LC-MS/MS). Based on different physical and chemical properties of these OPs metabolites, the solid phase extraction cartridges and the washing and eluting solvents were optimized in details. Furthermore, the mobile phase and mass spectrometric parameters were also investigated. The results showed that Oasis WAX cartridge was the best SPE column in this study, and 2 mL of NH3·H2O (5%) in methanol and 2 mL of methanol were chosen as the eluting solvents. The recoveries of six analytes were ranged from 60.5% to 104.0%, whereas DEP ranged from 17.8% to 36.2%. Seven analytes could be baseline separated from each other under the optimized chromatographic conditions. The limits of detection and quantification of seven analytes ranged from 0.005 to 0.2 μg/L and 0.02 to 0.5 μg/L, respectively. The standard deviations of response repeatability for intra-day and inter-day period were lower than 15.4%. This method was finally applied to determination of metabolites of OPs from 10 urines from general population in Guangzhou city. The concentrations of total OPs metabolites in urine samples ranged from 0.5 to 6.7 μg/L.
Liquid chromatography-tandem mass spectrometry; Organophosphate esters; Urine; Metabolites
21 June 2017; accepted 19 July 2017)
10.11895/j.issn.0253-3820.171027
