基于多特征融合预测蛋白质相互作用界面
2017-10-18陈心浩
陈心浩,胡 俭
(中南民族大学 生物医学工程学院,武汉 430074)
基于多特征融合预测蛋白质相互作用界面
陈心浩,胡 俭
(中南民族大学 生物医学工程学院,武汉 430074)
为高效准确地预测蛋白质相互作用界面,提取了传统特征,并采用多种方法改进进化信息特征,利用特征选择构建了一个14维的预测模型.通过5折交叉验证和独立测试,预测结果表明:该预测模型不仅显著降低特征维度,而且选择的特征组合具有较好的预测能力和较强的泛化能力.
蛋白质-蛋白质界面;分类;进化;特征选择
AbstractTo build a model of efficient and accurate classification of protein-protein interfaces, this study constructs two characteristics of traditional features and evolutionary information, a 14-dimensional feature model is constructed by feature selection.By cross-validation of the main data set and independent test set testing, results show that selects the features combination has better predictive ability and strong extension ability. Compared with the best models at the present stage, this study significantly reduce the dimensionality of the model case classification has improved.
Keywordsprotein-protein interface; classification; evolutionary; feature selection
区分蛋白质晶体中的生物学相互作用界面(Biological interfaces)和无生物学意义的晶体学界面(Crystal interfaces),是结构生物信息学中的一个重要研究方向.
现有计算方法预测蛋白质相互作用界面的特征主要分成两大类:第一类是以界面面积、疏水性和温度因子等几何特性和氨基酸理化特性为代表的传统特征[1];第二类则是以EPPIC方法为代表的进化特征[2].为获得良好的分类效果,目前的主要策略是将上述特征进行联合.然而,这类融合方法也存在弊端,如现阶段分类效果最好的Luo方法[3],该方法具有较高的特征维度(46维),且进化信息计算复杂,不利于快速构建本地分类模型.因此,本文期望采用较为简便的方式计算进化特征,融合传统特征并使用特征选择技术,构建一个低维高效的蛋白质互作界面分类模型.
1 材料和方法
1.1数据集,蛋白质界面残基、表面残基的定义
在构建和测试模型过程中使用了三个数据集,Duarte数据集[2]作为主数据集用于构建模型和优化参数,Bernauer[4]和Ponstingl[5]两个经典数据集作为独立测试集.
核心残基(Core)位于互作界面中心,主要由疏水性氨基酸构成.核心残基周围环绕着一圈残基,此类型残基称之为环绕残基(Rim).界面残基、表面残基、核心残基与环绕残基定义采用Proface方式定义[6].
1.2传统特征
核心残基(Core)与环绕残基(Rim): 分别计算核心残基与环绕残基在界面残基中的比例,构成Core和Rim 这两个特征.核心残基数目(NoC) :每个蛋白质复合体的核心残基数构成本特征.温度因子(BF):将PDB中每个残基温度因子做Z-score归一化,将归一化后的界面残基温度因子平均值作为此蛋白质复合体温度因子.局部包装密度( LD),热点残基数目(Nhs),氨基酸分布 (RP)定义方式来自Proface[6],界面疏水性 Hy)采用Jones定义[1].
1.3进化特征
本文采用默认参数,使用PSI-BLAST程序对目标蛋白质在NR数据库中搜索其同源序列并构建位置特异性矩阵.根据上述矩阵,采用Capra方法[7],对每一个残基位置分别计算了SE(Shannon entropy of residues),SERP(Shannon entropy of residue properties),VNE(von Neumann entropy),RE(Relative Entropy)和JSD(Jensen-Shannon divergence score)5种保守性分值,并且对于计算出来的5种保守性分值采用3窗口平均,构成另外5个保守性分值.计算公式如下:
SEi=-∑α∈AAp(α) lg[p(α) ] ,
(1)
SERPi=-∑α∈Term)p(β) lg[p(β) ] ,
(2)
VNEi=-Tr(ρlg(ρ) ) ,
ρ=diag[(p1,p2,…,p20)·BLUSUM62],
(3)
REi=-∑α∈AAp(α) lg[p(α)/q(α) ],
(4)
JSDi=λ∑α∈AAp(α) lg[p(α)/r(α) ]+
(1-λ) ∑α∈AAq(α) lg[q(α)/r(α) ],
(5)
WindowScorei=0.5Entropyi+

(6)
公式(1)中,p(α)是20种常见氨基酸在位置i出现的概率,公式(2)中的p(β)则是根据Mirny研究[8]对氨基酸根据化学属性分成6组,计算出的每一组在整体出现的概率,具体分组可见表1.VNE计算方法[9]如公式(3)所示,特点是将原始的概率得分使用BLUSUM62矩阵重新计算.RE的计算方式与SE接近,不同点是使用背景概率q(α)重新定义,其概率分布见表1.JSD是将RE做了背景频率改进[10],可以将保守性分数归一化0~1之间,在本文中λ=0.5.公式(6)即3窗口的算法,序列上第i个残基与其邻近的两个残基加权平均.将上述获得的5个保守性分值和5个窗口保守性分值分别作Z-score变换,以消除不同蛋白质复合体间差异.
蛋白质残基保守性分值可以衡量残基在进化过程中变异程度,生物学界面残基,特别是生物学界面上的核心残基在进化过程中相对保守.本文采用两种方式计算核心残基保守性分值[2],第一种是核心残基-界面残基保守性分值 (CI),计算核心残基保守性分值平均值与界面残基保守性分值平均值的差值,即将界面残基保守性分值作为基准.第二种是核心残基-表面残基保守性分值( CS),计算核心残基保守性分值平均值与表面残基保守性分值平均值的差值.最终构成20维进化信息特征.
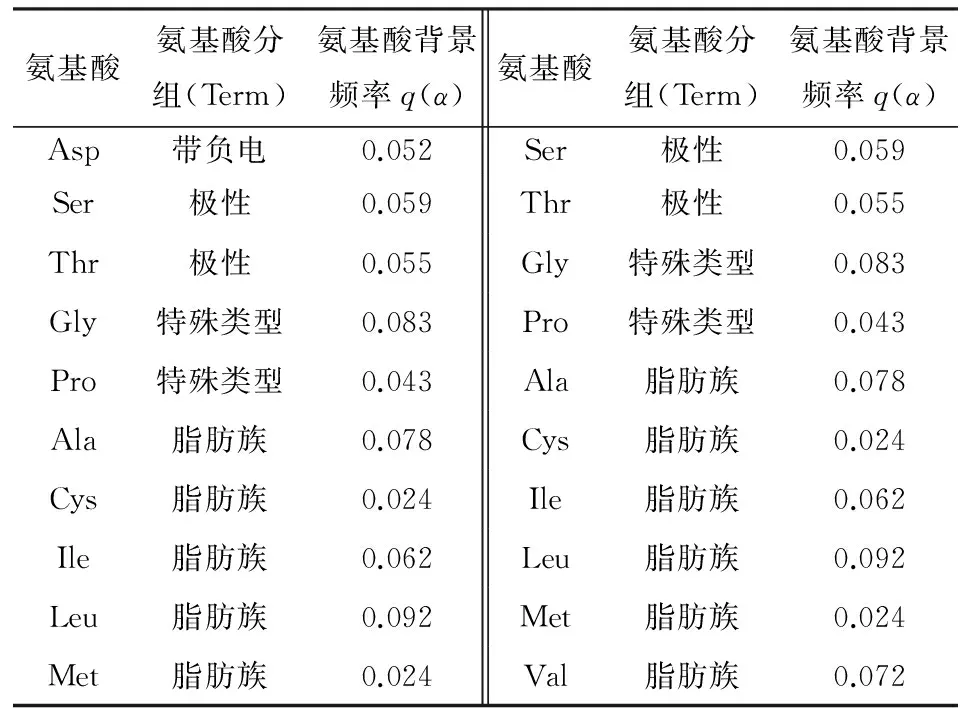
表1 氨基酸属性
1.4特征选择、分类器与分类评价
增L去R选择算法是一种改进了的前向特征选择方法[11].算法初始特征选择从空集开始,每轮先加入L维特征,然后从中除去R个特征,将每一轮AUC最高的特征组合挑选出来作为下一轮初始特征组合.
分类器采用R语言下随机森林包,所涉及参数均采用默认值.
对单个特征和联合特征测试均在Duarte数据集上完成,采用5折交叉验证.为排除随机影响,5折交叉验证采用50次独立分组取平均的结果,两个独立测试采用50次重复平均结果.分类效果评价采用敏感度(SN)、特异度(SP)、准确性、马修相关性系数(MCC)、受试者工作曲线(ROC)及ROC曲线下面积(AUC)6个指标.MCC范围是[-1, 1],当MCC大于0代表正确的分类效果,越接近1代表分类效果越好.一般来说,当MCC大于0.3表示有一定分类效果,大约0.5时分类效果较好.AUC也有类似的评价标准,当AUC处于0.5到0.6之间表示只有微弱的分类效果,当AUC大于0.6表示此特征有一定的区分样本能力,当AUC大约0.8表示分类效果很理想.
2 结果与讨论
2.1特征分类效果
根据表2,在传统特征中,Hy、Core、Rim、RP和Nhs5个特征的单独使用分类AUC均到达0.7以上,除Nhs每个特征的MCC都超过0.4,显示出这些特征在生物学界面和晶体学界面上有较大的分布差异性.BF和NoC的AUC处于0.6到0.7之间,MCC大于0.3,有一定分类效果.LD分类效果较差,AUC不到0.6.
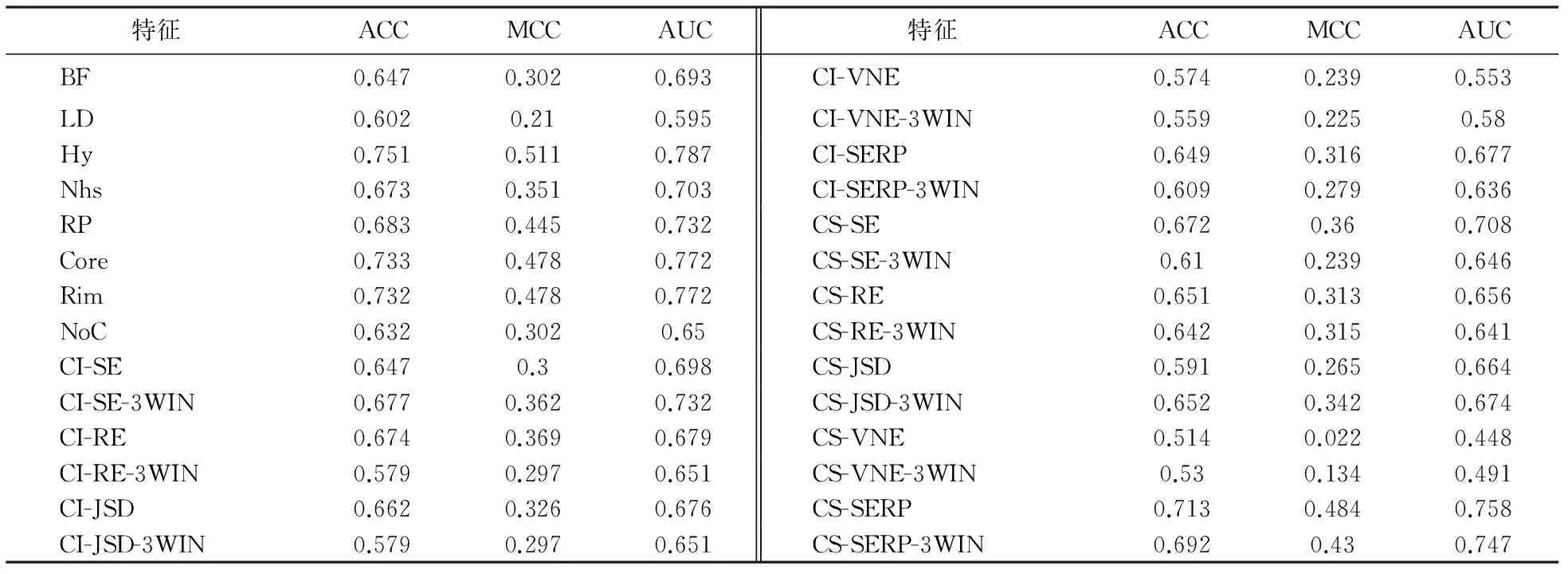
表2 特征独立使用分类效果Tab.2 Independent feature classification results
在进化信息特征中,并非所有的保守性分值算法都适合本问题,如CS-VNE的AUC小于0.5,产生相反的分类效果.若以AUC为评价准则,整体上来说,相同算法计算出的CS要略优于CI,这与Duarte得出的结论相同.在原始保守性分值与3维窗口计算出的保守性分数比较中,不同种算法产生了不同的效果,如CI-SE-3WIN相比CI-SE分类效果提升明显,而CS-SE-3WIN相比CS-SE分类效果却变差.在20个进化特征中,CS-SERPAUC达到0.758,MCC达到0.484,是28个特征中分类效果最好的特征之一.
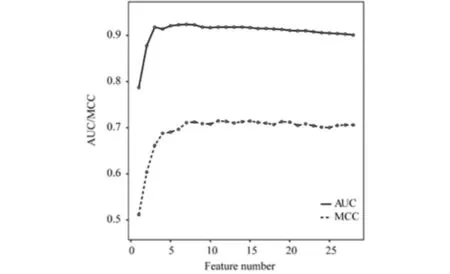
图1 特征选择Fig.1 Feature selection
2.2特征选择
以AUC为选择标准,本文采用增2去1选择算法对28个特征做特征选择.对于每一轮选择出的特征组合,计算AUC和MCC,绘制的曲线如图1所示,随着特征数目的增加,AUC先快速上升,在第8轮特征选择后达到顶点,而后AUC缓慢下降;MCC上升速度相比于AUC较慢,而且在达到顶点后并没有明显的下降趋势.综合AUC和MCC分值,最终选择第14个特征组合,分别是Hy、Core、CS-SERP-3WIN、CI-SE-3WIN、RP、Nhs、CI-SE、BF、CI-RE、CI-SERP、CI-JSD-3WIN、CI-RE-3WIN、CI-SERP-3WIN、LD.选择出的14个特征AUC为0.918,MCC为0.713,而全部28个特征AUC为0.901,MCC为0.706,可见本文在消减了一半特征维度情况下,AUC还是获得了较大程度提升,说明本文采用的特征选择确实可以在保证预测准确性条件下选择出更有意义的特征组合.
在特征选择中没有被选择出来的特征,其中Rim是因为与Core成对偶关系,所包含的信息是完全一致的;NoC是因为在本文中多个特征涉及到核心残基,信息上存在冗余因而没有被选择出来.信息冗余同样存在于20个进化信息特征上,因此只有8个进化信息特征被选择出来.虽然CS单个特征效果略好,但是在选择出的8个进化信息特征中只有一个CS,而独立使用LD分类效果较差却可以被选择出,说明并非联合较强特征一定会取得良好的分类效果,还需要考虑各个特征之间的组合效应.
2.3交叉验证与独立测试效果
表3所示的是Duarte数据集5折交叉验证结果和两个独立测试集的分类效果,图2所示的是相应的ROC曲线.可以看到,本文在Duarte数据集上取得了AUC为0.918,MCC为0.713这样良好的分类效果.将本方法应用于两个独立测试集上,Bernauer数据集AUC达到0.955,MCC达到0.745的MCC,Ponstingl数据集AUC为0.962,MCC为0.842,均获得了良好的的分类效果,可见本方法有较强的泛化能力.
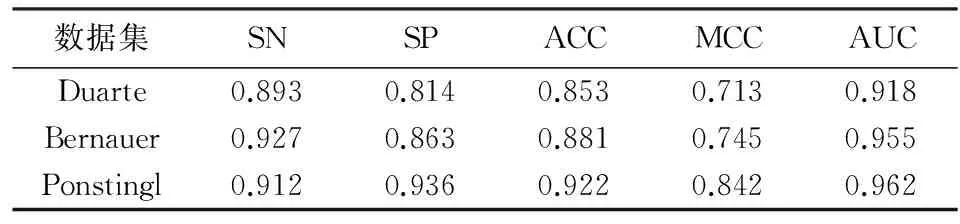
表3 Duarte数据集5折交叉验证和独立测试集预测效果
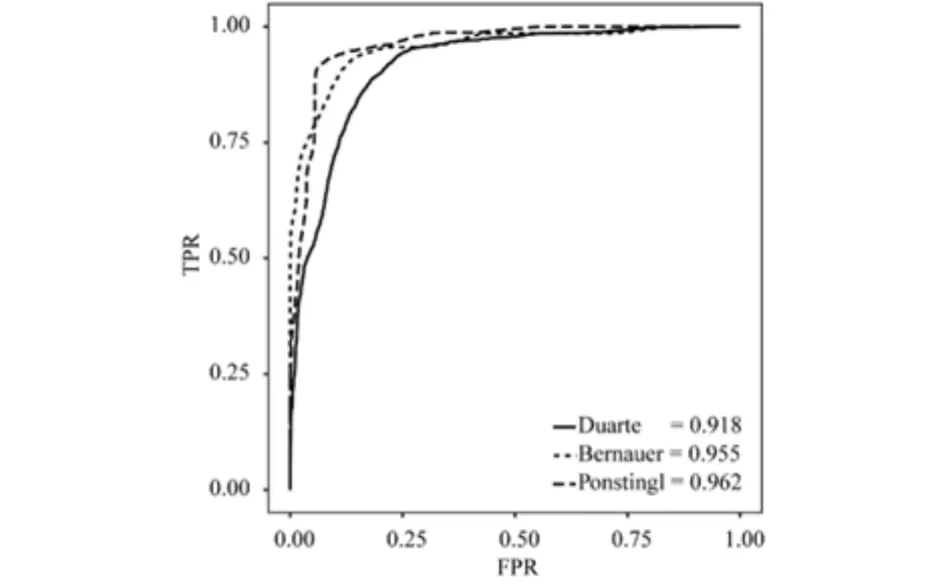
图2 Duarte数据集5折交叉验证和独立测试集ROC曲线Fig2 The ROC curves of 5-fold cross validation test and two independent datasets
2.4与现有方法比较
为更加全面地评价本方法,本文采用现阶段分类效果最好的两个分类器,即Luo方法和EPPIC方法对Duarte数据集做5折交叉验证,与本方法得到的结果进行比较.EPPIC方法的预测效果直接取自文献报道;对Luo使用的特征数据,采用与本文相同的50次5折交叉验证进行评价.本方法与这两种方法比较见图3,从对比结果上来看,除SN本方法与现有方法相仿之外,SP、ACC和MCC本方法均有显著提升,采用符号秩和检验SP、ACC和MCC本方法差异达到5.24E-10、3.01E-09、5.19E-09,可以得出本方法在Duarte数据集上分类效果优于上述两种方法的结论.
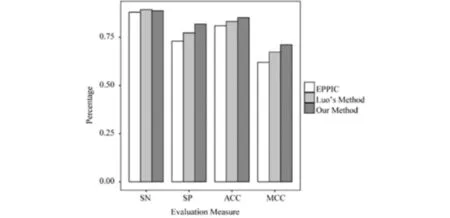
图3 本文方法与Luo方法、EPPIC比较Fig.3 Comparison of the performances of our method and Luo's Method and EPPIC
3 结语
本文提取了进化特征和传统特征,通过特征选择构建了一个高效的蛋白质相互作用界面分类模型.交叉验证和独立测试的结果表明本方法可以达到较为理想的预测效果.与现有方法相比,本方法大幅度降低了特征维度,却并没有降低分类效果.然而也有不完善的地方,如备选特征数目较少,对特征的生物学意义挖掘不深等,这些问题将是作者下一步研究的重点.
[1] Jones S,JM Thornton. Analysis of protein-protein interaction sites using surface patches[J]. Journal of Molecular Biology, 1997, 272(1): 121-132.
[2] Duarte J M, Srebniak A, Scharer, M A, et al. Protein interface classification by evolutionary analysis[J]. BMC Bioinformatics, 2012, 13(1): 334-334.
[3] Luo J, Guo Y, Fu Y, et al. Effective discrimination between biologically relevant contacts and crystal packing contacts using new determinants[J]. Proteins, 2014, 82(11): 3090-3100.
[4] Bernauer J, Bahadur R P, Rodier, et al. DiMoVo: a Voronoi tessellation-based method for discriminating crystallographic and biological protein-protein interactions[J]. Bioinformatics, 2008, 24(5): 652-658.
[5] Ponstingl H, Kabir T, Thornton J M. Automatic inference of protein quaternary structure from crystals[J]. Journal of Applied Crystallography, 2003, 36(5): 1116-1122.
[6] Saha R P, Bahadur R P, Pal A, et al. ProFace: a server for the analysis of the physicochemical features of protein-protein interfaces[J]. BMC Struct Biol, 2006, 6: 11.
[7] Capra J A, Singh M. Predicting functionally important residues from sequence conservation[J]. Bioinformatics, 2007,23(15): 1875-1882.
[8] Mirny L A, Shakhnovich E I. Universally conserved positions in protein folds: reading evolutionary signals about stability, folding kinetics and function[J]. Journal of Molecular Biology, 1999, 291(1): 177-196.
[9] Caffrey D R, Somaroo S, Hughes J, et al. Are protein-protein interfaces more conserved in sequence than the rest of the protein surface[J]. Protein Science, 2004, 13(1): p. 190-202.
[10] Lin J, Divergence measures based on the Shannon entropy[J]. IEEE Transactions on Information Theory, 1991,37(1): 145-151.
[11] 姚 旭, 王晓丹, 张玉玺, 等. 特征选择方法综述[J]. 控制与决策,2012,27(2):161-166.
StudyonProtein-ProteinInterfacialClassificationBasedonMulti-featureFusion
ChenXinhao,HuJian
(College of Biomedical Engineering, South-Central University for Nationalities, Wuhan 430074, China)
Q811.4
A
1672-4321(2017)03-0080-04
2017-03-30
陈心浩(1968-),男,副教授,研究方向:医学图像处理与传输,E-mail: xinhaochen@mail.scuec.edu.cn
国家自然科学基金资助项目(61002046);中央高校基本科研业务专项基金项目(CZP17025)
