白念珠菌几丁质合成酶三维结构的同源建模及其与FR-900403的分子对接研究
2017-09-18孙彬刘敏赵世振王世本黄宏丽
孙彬 刘敏 赵世振 王世本 黄宏丽
(1.聊城大学生物制药研究院,聊城 252000;2.沈阳药科大学制药工程学院,沈阳 110016)
·论著·
白念珠菌几丁质合成酶三维结构的同源建模及其与FR-900403的分子对接研究
孙彬1刘敏1赵世振2王世本1黄宏丽1
(1.聊城大学生物制药研究院,聊城 252000;2.沈阳药科大学制药工程学院,沈阳 110016)
目的 探究白念珠菌几丁质合成酶活性位点的结构特征。方法 通过采用同源建模的方法首次构建白念珠菌几丁质合成酶的三维结构模型,模型的可靠性经Ramachandran和Profile-3D图进行验证。采用InsightⅡ-Binding site方法准确定位几丁质合成酶的活性位点,并研究了几丁质合成酶的重要功能残基在活性位点的立体分布。结果 通过柔性分子对接方法首次阐明几丁质合成酶抑制剂FR-900403与靶酶活性位点的相互作用模式,明确几丁质合成酶与该类抑制剂结合时起重要作用的氨基酸残基。结论 本研究为基于几丁质合成酶三维结构的药物靶点设计提供重要的参考信息,同时也为抗真菌药的发展奠定坚实的理论基础。
真菌感染;几丁质合成酶;同源建模;分子对接
近年来,随着广谱抗生素、免疫抑制剂的广泛和长期使用以及艾滋病和癌症放疗化疗患者的日渐增多等因素,侵入性真菌感染发病率逐年攀升[1]。研究发现引起侵入性真菌感染的最常见致病菌包括念珠菌属 (约占侵入性真菌感染病例的70%~90%)和曲霉菌,这些致病真菌在这些免疫系统受损的患者中会引起很高的死亡率,目前此类感染已成为重要的健康问题[2-4]。当前,临床上应用的抗真菌药物主要包括:作用于真菌细胞膜,损害细胞膜脂质结构和功能的多烯类如两性霉素B;作用于真菌细胞P450的唑类化合物;作用于角鲨烯环氧化酶的丙烯胺类以及干扰真菌核酸合成的嘧啶类化合物等,但这些抗真菌药物均有较为明显的毒副作用,并且随着临床上预防以及治疗上的大量使用,已出现越来越多的耐药菌[5-6]。真菌细胞壁是真菌细胞所独有的结构,不存在于哺乳动物细胞中,其作用是控制细胞内压力以维持菌体的完整性;几丁质是构成细胞壁的重要成分之一,一旦几丁质的合成出现问题,则会导致细胞壁的缺失破损,从而引起真菌细胞的膨胀破裂死亡[7-8]。几丁质合成酶抑制剂能够以几丁质酶为靶点,通过发生特异性的结合来抑制几丁质合成酶的生物活性,阻断其真菌增殖的过程,从而起到抗真菌的作用。因此研究几丁质酶的活性位点以及与几丁质酶抑制剂的相互作用方式对致病真菌的治疗具有重要意义。
本文中选取了一种代表性的致病真菌——念珠菌种属中的白念珠菌 (Candidaalbicans)作为研究对象,探究其几丁质合成酶的结构特点以及与几丁质合成酶抑制剂的相互作用方式。但是其三维结构至今尚未被解析出来,阻碍了对该酶的功能、催化机理以及基于靶点的几丁质合成酶抑制剂的进一步研究。同源建模和分子动力学模拟的方法能很好地应用于生物大分子的理论模拟,并指导和解释实验现象。本文采用这两种方法构建白念珠菌几丁质合成酶的三维结构模型,并通过分子对接和量子化学计算方法考察几丁质合成酶抑制剂FR-900403[9]与白念珠菌几丁质合成酶的结合模式,以期为基于念珠菌属几丁质合成酶的三维结构药物设计提供重要的参考信息。
1 材料与方法
1.1 仪器与试剂
所有同源建模、模型优化及分子对接计算均在Dell Precision工作站上完成,所用程序为SYBYL-Modeller[10],Discovery Studio Visualizer client,CHARMm,Simulation protocols和AutoDock[11-12],计算中选用的各项参数除特别说明外均使用缺省值。
1.2 同源建模
在瑞士生物信息研究所的“蛋白专家分析系统”(www.expasy.org)中用“Chitin synthase”作关键词搜索Swiss-Prot/TrEMBL数据库得到白念珠菌的几丁质合成酶一级结构 (序列号:P23316)[13],它是一个包含776个氨基酸的蛋白,其序列见图1。
以白念珠菌的几丁质合成酶 (Chitin synthase)一级序列 (来源于UniProtKB/Swiss-Prot数据库,编号P23316)为探针,在UniProt数据库中利用BLAST-P程序搜索Protein Date Bank (PDB)蛋白晶体数据库中的同源性的模板蛋白的三维结构。整个模型的构建分为以下两个步骤。首先利用Clustal X2程序进行序列比对,确定模板与序列之间的残基匹配。再进行模型的建立:根据序列比对的结果,采用Modeler自动建模程序搭建白念珠菌合成酶的三维结构并进行侧链异构化,对所得模型在pH为8.0条件下进行加氢、补键等化学修饰,然后把模板蛋白中的配体化合物赋到建模所得结构的相应位置,得到目标蛋白的初始结构模型。
1.3 分子动力学模拟和能量优化
利用Modeler的方法构建完几丁质合成酶模型后,对其进行了分子力学和分子动力学的优化。首先在Macromolecules的模块下的Prepare Protein操作界面上的优化残基侧链,并在指定的pH值下质子化结构。随后利用Simulation/Dynamics文件夹下的Solvation流程,对其结构进行溶剂化。Add Counterion选项选择Salt溶液类型。Salt Concentration采用缺省值0.145,阳离子类型设置为Sodium,即Na+,阴离子类型 (Anion Type)设置为Chloride,即Cl-。以上操作表示为复合物添加的溶剂环境为生理盐水。并对蛋白-配体复合物体系进行束缚,最后进行的Standard Dynamics Cascade流程通过优化I (Minimization)、优化II (Minimization2)、升温 (Heating)、平衡 (Equilibration)、模拟采样 (Production),这5个阶段的方法进行计算。
1.4 分子对接
采用SYBYL分子片段库中片段构建FR-900403分子结构。SYBYL/MINIMIZE,Tripos标准分子力场,Gasteiger-Hückel电荷。Powll能量优化方法,能量收敛标准为0.042 kJ·mol-1·nm-1。首先对化合物进行分子动力学模拟退火,参数设置:升温到1 000 K,平台处理时间为1.0 ps,退火至250 K,退火时间为1.5 ps,指数型退火函数。Tripos标准分子力场,Gasteiger-Hückel电荷,每次循环,得50个构象并能量优化,找到能量最低的构象作为下一步的起始构象。反复操作,以重复出现5次以上的最低能量构象作为该分子的最低能量构象,用于下一步的柔性对接。
柔性对接采用AutoDock程序,它能够自动完成配体和受体的对接,对给定的由配体分子和受体分子组成的体系,能自动优化小分子化合物的取向及构象,建立靶标的优化模型,寻找小分子与靶标大分子作用的结合位点和最佳结合构象,计算其与生物大分子的相互作用能来选择最优匹配模式。
2 结 果
2.1 序列比对结果
当我们构建蛋白序列为白念珠菌的几丁质合成酶3D模型时,首先需要挑选一个或多个合适的同源模板 (templates)。根据几丁质合成酶的一级结构,采用序列相似性搜索程序BLAST从己知的蛋白结构中识别出同源性较高的一个或多个模板蛋白。进行BLAST搜索时,使用蛋白数据库 (Protein Data Bank,PDB)来寻找模板蛋白,之后得到打分较高的模板蛋白,如表1所示。
一个理想的模板蛋白首先需要具有较高的序列同源性 (Sequence identity),并且V-value要足够小 (<1×10-5)。通过对几丁质合成酶结构域序列比对结果进行分析发现,此类结构显示出高度序列同源性,说明该家族具有相似的二级结构和三级结构 (见图1)。来自同丝水霉的Chitin synthase 1 (SAPMO,PDB code:2MPK)和酵母菌的Chitin synthase 3 (YEAST,PDB code:4WJW)在BLAST比对结果显示出具有32.7%和31.9%的一致性序列同源性 (Sequence identity),且打分最高,E-value均相对合适。进一步的研究发现,来自同丝水霉的Chitin synthase 1的蛋白晶体结构2MPK仅解析了其中A链的74个氨基酸残基,相对较少。而来自酵母真菌几丁质合成酶3的蛋白晶体结构4WJW同源性同样大于30%,氨基酸残基较为接近,并且分辨率较高。因此,最终选择酵母真菌的Chitin synthase 3作为Chitin synthase的同源模板序列。采用Align Sequence to Templates工具将这这个模板序列和白念珠菌-几丁质合成酶的序列进行了多重序列比对,参数设置选择默认值。多重序列比对结果见图2。
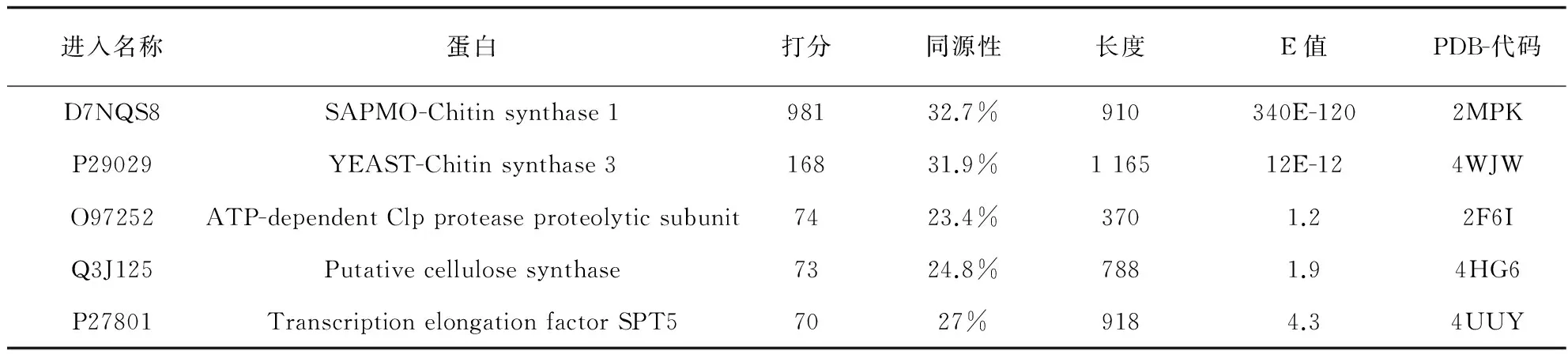
表1 白念珠菌几丁质合成酶的一级结构比对结果
比对完全相同的氨基酸残基用蓝色进行了标注,深蓝色代表保守区域,浅蓝色代表半保守区域。通过对白念珠菌几丁质合成酶的序列比对结果分析发现:在几丁质合成酶的一级结构序列67PDTFSAEGFTL77、104ARTMHGVMKN113、147AVVDGFDFD155、157SVLELLTAT165、198ARTMHGVMKNSIDENLKFKGDEK206、218SVLELLTATKESNQKKINSH228、467SVLELLTATKESNQKKINSHLFSFFSLSNFYLT479和499VSYKSLVSYKSLG505上分别对应着活性位点上的关键氨基酸残基,并且这些关键氨基酸残基与YEAST-Chitin synthase 3具有这些较高的序列同源性。由此可知,这两类蛋白酶的活性位点区域高度保守,很可能具有相近的空间构型,从而为白念珠菌几丁质合成酶同源模型的构建奠定了理论基础。
2.2 同源建模结果
由自动建模程序Modeler构建的初始模型,经过能量最小化和分子动力学模拟后,得到了白念珠菌几丁质合成酶三维结构的合理模型,如图3所示,Chitin synthase的三维结构中含有22个α螺旋、11个β折叠和12个γ转角。在分子动力学的模拟过程中,蛋白质体系在12 ns之后达到收敛,RMSD值也在最后达到了平衡,所以选择10 ns到12 ns的结构作为最终结构模型。并通过在线结构评估软件 (http://nihserver.mbi.ucla.edu)进行评估,在评价结果中主要考察所有氨基酸残基骨架的二面角分布,并通过程序生成的拉氏图 (Ramachandran)来表示 (见图4a)[14]。建模的几丁质合成酶模型有95.5%的残基落入最佳区域 (The most favoured region),3.4% 的残基落入其他许可区 (Additional allowed regions),0.5%的残基落入勉强许可区 (Generously allowed regions),0.6%的残基落入不允许区 (Disallowed regions)。上述结果说明,建模所得几丁质合成酶模型的氨基酸二面角结构是比较合理的,同源模型中的氨基酸残基Cys14、Glu210、Ser346、Val376、Phe468和Asn518虽处于不可接受区,但是这些氨基酸残基大多都处于模型的表面,远离活性位点,对下一步的活性位点的对接研究没有太大的影响。之后采用Profile-3D (见图4b)检测建模蛋白氨基酸序列与其三维结构的兼容性,发现所有残基的分值均在0.10以上,无不兼容残基。以上检测结果表明,建模所得的几丁质合成酶三维结构是合理可信的。
2.3 抑制剂FR-900403与白念珠菌几丁质合成酶活性位点的相互作用模式

图1 白念珠菌几丁质合成酶的氨基酸序列 图2 白念珠菌几丁质合成酶的氨基酸序列与酵母真菌Chitin synthase 3序列 (4WJW)进行同源性比对 (深蓝.保守序列,浅蓝.半保守序列) 图3 通过同源建模构建的白念珠菌几丁质合成酶3D结构 图4 a.利用Ramachandran plot评估几丁质合成酶模型结构,b.利用Profile-3D plot评估几丁质合成酶模型结构
Fig.1 The amino acid sequence ofCandidaalbicanschitin synthase Fig.2 Sequence alignments ofCandidaalbicanschitin synthase and YEAST-Chitin synthase 3 (4WJW) with conserved (constitution,deep blue;semiconserved constitution,blue) Fig.3 3D models ofCandidaalbicanschitin synthase generated by Modeler Fig.4 a.Ramachandran plot of Chitin synthase 3D-structure modeling,b.Profile-3D plots of the Chitin synthase 3D-structure modeling
从Kemiasp.中分离出含有嘌呤和四氢呋喃环的天然产物FR-900403是白念珠菌几丁质合成酶的有效抑制剂 (见图5),它在体外的MIC为0.4 μg/mL,并且具有一定的体内活性。研究FR-900403与白念珠菌活性位点的相互作用模式有助于阐明FR-900403这类几丁质合成酶酶抑制剂的分子作用机理,为进一步的结构优化指明方向,也可以为开展全新药物设计提供思路。
在同源建模所得几丁质合成酶的三维结构基础上,利用Binding-site程序自动生成几丁质合成酶最可能的活性位点,主要由Lys243、Pro244、Asp245、Asn246、Val285、Ala286、Ser287、Gln288、Asn289、Phe464、Tyr465、Leu535等氨基酸残基组成,并且这些关键氨基酸残基大多处在序列比对的保守区域中,因此选择该活性位点作为下一步分子对接研究的基础。
几丁质合成酶抑制剂FR-900403与白念珠菌几丁质合成酶的对接结果见图6,Csore的综合评分为4.95分,表明此次对接结果较为可信。从图6的对接结果可以看出,FR-900403与建模蛋白几丁质合成酶的主要作用是由2个区域组成:一是由Phe464和Tyr 465构成的疏水性结合腔,而FR-900403上的嘌呤环基团主要结合在由Phe464和Tyr465构成的疏水性结合活性腔这一部位,并与其中的关键氨基酸Tyr 465形成π-π共轭疏水相互作用,这表明嘌呤环基团是几丁质合成酶抑制剂FR-900403的必需基团,将该基团用其他基团替代后会大大降低的抑制活性;二是几丁质合成酶活性腔中的酸性氨基酸Asp245、极性氨基酸Asn246和Asn289可以形成亲水结合腔,而抑制剂FR-900403上的四氢呋喃环以及支链部分则可以与该区域形成4对氢键相互作用;由于氢键相互作用是抑制剂与靶酶蛋白结合的基本作用力,这可能是该类化合物产生高抑制活性的重要分子基础。
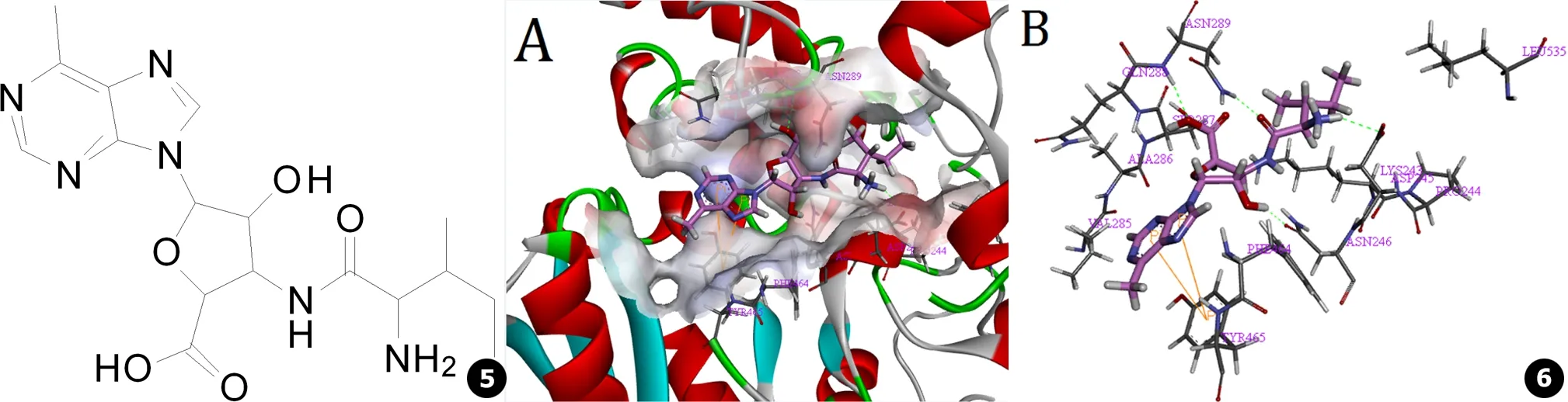
图5 FR-900403的分子结构 图6 抑制剂FR-900403与白念珠菌几丁质合成酶的对接结果
3 讨 论
本研究以来自酵母菌几丁质合成酶的晶体结构为模板,采用同源建模和分子动力学方法首次建模了白念珠菌几丁质合成酶的三维结构,建模结构的精度经Ramachandran图和Profile-3D图进行了验证,其结果都表明几丁质同源模型的合理性。接着,对几丁质合成酶活性腔进行分析发现,该部分主要由Lys243、Pro244、Asp245、Asn246、Val285、Ala286、Ser287、Gln288、Asn289、Phe464、Tyr465、Leu535等关键氨基酸残基组成,这些氨基酸构成了活性腔的空间结构特征。利用已发现的几丁质合成酶抑制剂FR-900403与靶酶进行分子对接,从对接结果中可以看出几丁质合成酶活性腔中的酸性氨基酸残基Asp245和极性氨基酸残基Asn246、Asn289均可以与FR-900403结构中的四氢呋喃环以及支链部分形成氢键作用;同时Tyr465可以与FR-900403结构中嘌呤环部分形成π-π共轭作用,这些弱力相互作用增强了抑制剂与靶酶的结合能力。本次对接的结果在分子水平上揭示了几丁质合成酶抑制剂FR-900403与靶酶受体的结合方式。该项研究工作对于下一步针对抗真菌几丁质合成酶抑制剂结构改造和优化指明了方向,同时也为抗真菌新药的发现打下了基础。
致谢:感谢抗体药物协同创新中心及纳米药物与释药系统工程中心提供的帮助。
[1] 余靓平,谢小云,魏文树,等.抗深部真菌感染药物的特性及临床应用[J].中南药学,2012,10(5):380-384.
[2] 潘伟华.抗真菌药物的应用与发展[J].世界临床医药,2014,35 (12):705-708.
[3] Groll AH,De Lucca AJ,Walsb TI.Emerging targets for the development of novel antifungal therapeutics[J].Trends Mierobiol,1998,6 (3):117-124.
[4] 戴振国,徐文泉,董华军.抗真菌药的分类及临床应用[J].中国医刊,2007,42(2):58-61.
[5] Silveira FP,Husain S.Fungal Infections in solid organ transplantation[J].Am J Transplant,2013,13(4):162-271.
[6] Richardson MD.Changing patterns and trends in systemic fungal infections[J].J Antimicrob Chemother,2005,56(Suppl1):5-11.
[7] Xie J,Thellend A,Becker H,et al.Synthesis and evaluation of a C-glycosyl nucleoside as an inhibitor of chitin synthase[J].Carbohydr Res,2001,334(3):177-182.
[8] Kim MK,Park HS,Kim CH,et al.Inhibitory effect of nikkomycin Z on chitin synthases inCandidaalbicans[J].Yeast,2002,19(4):341-349.
[9] Pfefferle W,Anke H,Bross M,et al.Asperfuran,a novel antifungal metabolite fromAspergillusoryzae[J].J Antibiot,1990,43(6):648-654.
[10] Fiser A,Sali A.Modeller:generation and refinement of homology-based protein structure models[J].Method Enzymol,2003,374 (374):461-491.
[11] 陈凯先,蒋华良,嵇汝运.计算机辅助药物设计——原理、方法及应用[M].上海科学技术出版社,2000:256-281.
[12] 徐筱杰,侯廷军,乔学斌等.计算机辅助药物分子设计[M].化学工业出版社,2004:325-342.
[13] Boeckmann B,Bairoch A,Apweiler R,et al.The Swiss-Prot protein knowledgebase and its supplement TrEMBL in 2003[J].NucleicAcids Res,2003,31(1):365-370.
[14] Sali A,Blundell TL.Comparative protein modelling by satisfaction of spatialrestraints[J].J Mol Biol,1993,234 (3):779-815.
[本文编辑] 卫凤莲
Homology modeling ofCandidaalbicanschitin synthase and its molecular docking study with FR-900403
SUN Bin1,LIU Min1,ZHAO Shi-zhen2,WANG Shi-ben1,HUANG Hong-li1
(1.InstituteofBioPharmceuticalResearch,LiaochengUniversity,Liaocheng252000,China;2.SchoolofPharmaceuticalEngineering,ShenyangPharmaceuticalUniversity,Shenyang110016,China)
Objective In order to further explore the structural features of the active site ofCandidaalbicanschitin synthase.Methods Homologous 3D model ofCandidaalbicanschitin synthase was built on the basis of the crystal coordinates of Yeast Chitin synthase,while the reliability of the model was assessed by Ramachandran plot and Profile-3D analysis.The active site ofCandidaalbicanschitin synthase was searched by Insight II-binding site analysis and important functional residues were located at the active site.To explore the binding mode of the Chitin synthase inhibitors FR-900403 with the active site ofCandidaalbicanschitin synthase,FR-900403 was docked into the active site.Results The binding pattern predicted by the affinity module revealed that important residues interacted with the inhibitors FR-900403.Conclusion The study provided further refinement of the chitin synthase inhibitor binding interaction that may be used as a basis for new structure-based design efforts and discovery of antifungal agents.
fungal infections;chitin synthase;homology modeling;molecular docking
193-197]
国家自然科学基金 (81703357),山东省自然科学基金 (ZR201702060112)
孙彬,男 (汉族),博士,讲师.E-mail:goengoy@163.com
R 379.4
A
1673-3827(2017)12-0193-05
2016-11-17
