全氟温室气体C2F6与H自由基脱氟反应机理研究
2017-06-29刘艳,于虹
刘 艳 ,于 虹
(1.渭南师范学院 化学与材料学院,陕西 渭南 714099;2.陕西师范大学 化学化工学院,西安 710062)
【现代应用技术研究】
全氟温室气体C2F6与H自由基脱氟反应机理研究
刘 艳1,2,于 虹1
(1.渭南师范学院 化学与材料学院,陕西 渭南 714099;2.陕西师范大学 化学化工学院,西安 710062)
采用密度泛函B3PW91方法,在6-311++g(d,p)基组水平研究了全氟气体C2F6与H自由基的脱氟反应机理。通过振动频率和内禀反应坐标(IRC)分析证实了各过渡态的准确性,获得了3条可能的反应通道。结果表明:C2F6+H→TS2→C2F5+HF是最佳的反应通道,生成的产物C2F5+HF的能量低于反应物C2F6+H的能量,是放热反应,该反应通道需要克服的能垒高度为130.6 kJ/mol,是三条反应通道中最低的,不管从动力学还是热力学角度上分析都是最有利的。
C2F6;H自由基;反应机理
全氟气体(PFCs)是一类人造强温室气体,常见于半导体的制造和铝业生产的废气中。其中,C2F6具有无色、无味、较易液化、微溶于水和稳定性高等特性[1-2],具有高的使用价值。但在温室效应上,C2F6的危害大约是等量CO2的11 900倍。C2F6分子中强共价键C-F键的平均键能高达485.3 kJ/mol[3],这使得C2F6非常稳定而不容易被降解,在大气层中的生命周期长达一万年,并且此效应具有累计的不可逆性。因此,研究如何消除温室气体C2F6,进而阻止其对地球温度的影响具有重要意义。有关C2F6气体反应的相关文献较少,但与C2F6类似的含氟类强温室气体SF6CF3相比,已有较多文献报道SF6CF3不能与氧化性自由基发生反应或反应很慢[4-6],但可与还原性自由基(H·[7]、CH3·和C2H3·[8])发生反应。而C2F6与还原性自由基,例如H自由基的反应目前还未有实验或理论方面研究报道。本文将从理论上采用密度泛函理论(DFT)研究气相中C2F6与还原性自由基H的微观反应机理及可能产物,预测C2F6与还原性自由基间可能存在的降解途径,为C2F6在大气中的汇提供必要的理论依据,并对含氟类化合物的治理提供理论指导。
1 计算方法
用密度泛函[9-10]B3PW91/6-311++g(d,p)计算方法,优化了C2F6与还原性自由基H反应路径上的各反应物、过渡态和产物等物种的几何构型,通过振动频率分析对稳定点和过渡态结构予以确认。在相同水平上用内禀反应坐标[11-12](IRC)方法确认各过渡态与相应的反应物、中间体或产物关联性,即各个过渡态都准确地连接到了相应的反应物和产物两端,计算工作均利用 Gaussian 09[13]程序完成。用传统过渡态理论计算了各反应通道在200~2 000 K温度范围内的速率常数。
2 结果与讨论
B3PW91/6-311++g(d,p)水平优化得到的反应物、过渡态和产物的几何构型参数及部分实验值见图1。反应物 C2F6中 C-C和C-F键长分别为 0.155 4 和0.133 1 nm,键角 F-C-F为109.0°,实验所得数据分别为0.156 nm、0.132 nm和109.5°[14]。因此,理论计算获得的结果与实验值数据相差很小,证明所选用B3PW91/6-311++g(d,p)计算方法是可靠的。图2为C2F6+H反应体系在B3PW91/6-311++g(d,p)计算方法下获得的势能剖面图(以反应物C2F6+H的能量作为能量参考点)。表1为所有反应物、过渡态和产物在B3PW91/6-311++g(d,p)水平上的振动频率,从表中的振动分析结果可以看出,各反应物和产物的振动频率均是正值,为反应势能面上的稳定点,而过渡态都有且仅有唯一的虚频率。对三个过渡态进行IRC计算结果显示:其振动分别指向三条通道各自的反应物和产物两端,是势能面上的一阶鞍点。表2为C2F6+H反应体系中各物种的电子结构能(U)、零点能(ZPE)及总能量(ET)和相对能量(ER)。
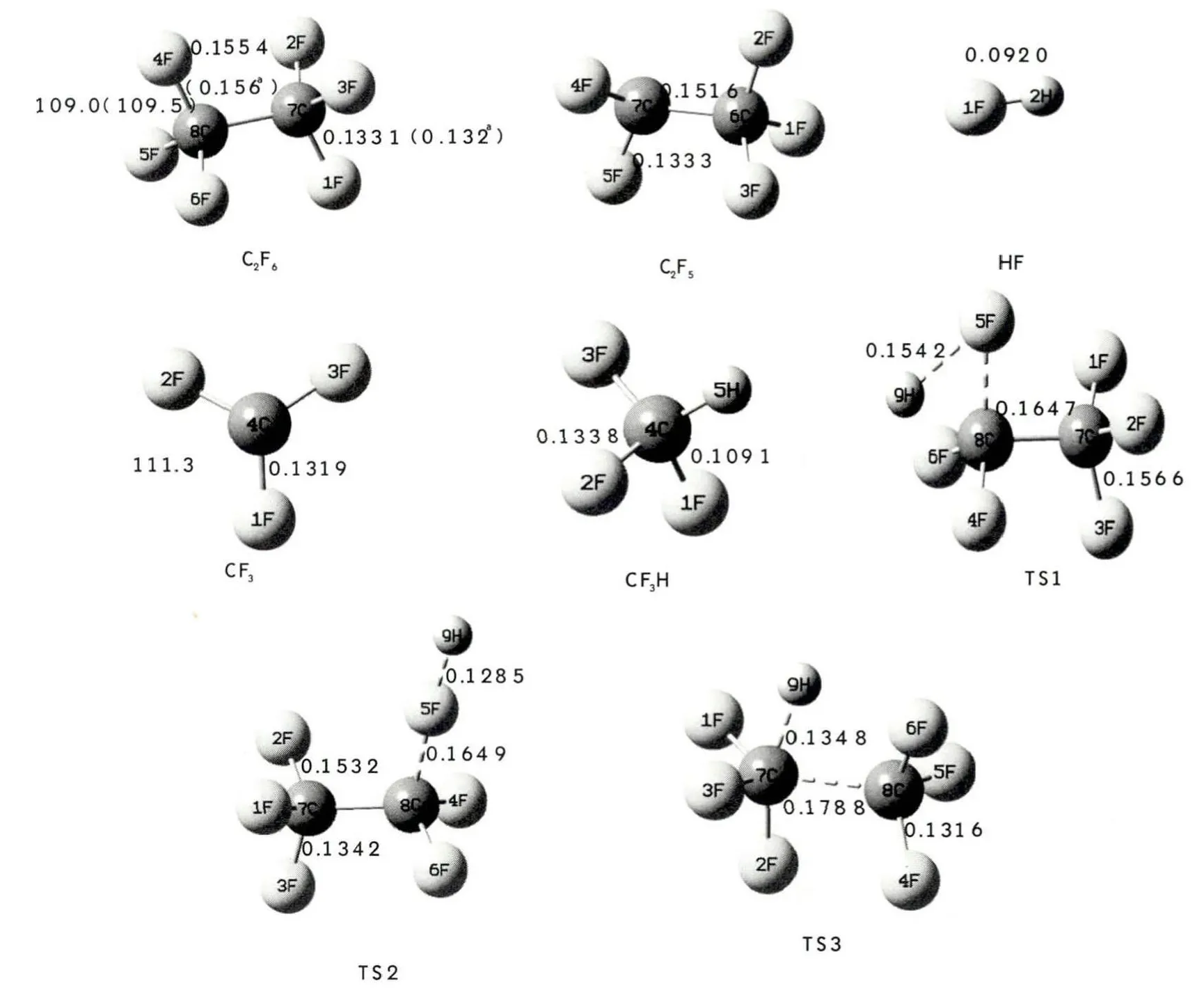
图1 B3PW91/6-311++g(d,p)水平优化得到的各物种的几何构型(键长:nm;键角:°;指数a是来自文献[14]的实验数据)
2.1 反应机理讨论
C2F6与H自由基可经过3条不同的反应通道(通道 1~3)生成4种小分子产物C2F5、HF、CF3和CF3H,为方便讨论,分别标记P1为(C2F5+ HF), P2为(CF3+CF3H)。其中,通道 1和通道 2生成相同产物P1,通道 3生成产物P2。反应流程如下:
C2F6+ H→TS1→P1(C2F5+ HF),(通道1)
C2F6+ H→TS2→P1(C2F5+ HF),(通道2)
C2F6+ H →TS3→P2(CF3+CF3H)。 (通道3)
3条反应通道分别对应3种反应机理:(1) H自由基插入C2F6分子中的C-F键之间,致使C-F键断裂;(2) H自由基与C2F6中的F原子发生相互抽提反应;(3) H自由基插入C2F6分子中的C-C单键之间,致使C-C键断裂。
通道1中,H原子靠近C2F6,进攻其中一个 F原子,由于两个C原子相连的六个F原子所处位置相同,因此无论替换哪个F原子所得到的情况都是相同的,形成过渡态TS1。TS1中,欲断裂的C-F 键长由原来的0.133 1 nm拉长到0.164 7 nm,欲形成H-F键,其长度缩短至0.154 2 nm。TS1的虚频值为-1 207.4 cm-1,虚频的振动合运动主要表现为C(8)-F(5)键的伸缩振动,并伴随H原子的靠近。可见,该过渡态虚频的振动模式与相应过渡态走向反应物或产物的位移向量相对应。TS1的能垒高度相对于反应物为228.6 kJ/mol。接着,H原子携带F原子离去生成裂解产物P1 (HF+C2F5),同时释放出293.7 kJ/mol的能量,因此,该反应为放热反应。
通道 2中,H原子直接进攻C2F6中的F原子,发生直接抽提反应。反应过程中,F(5)H(9)间距离不断减小,而C(8)F(5)键长逐渐拉长,其余化学键基本保持不变。当F(5)H(9)距离由无限远离状态减小至0.128 5 nm,C(8)F(5)的距离由0.133 1 nm拉长到0.164 9 nm时,形成的过渡态TS2,其虚频值为-1 537.2 cm-1。形成TS2需要跨越的势垒高度为130.6 kJ/mol,比TS1的能垒低98.0 kJ/mol。之后,TS2释放出195.7 kJ/mol的能量,形成产物P1,其能量比反应物低-65.1 kJ/mol。通道 2同样为放热反应,但释放的热量比通道 1少了98.0 kJ/mol。从动力学考虑,相对于通道 1,通道 2更易发生。
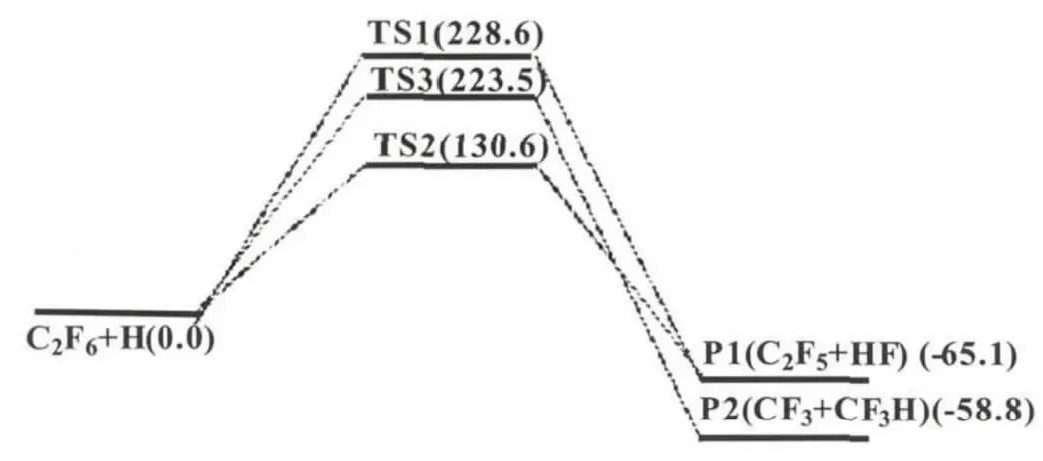
图2 B3PW91/6-311++g(d,p)水平上反应势能剖面图(能量:kJ/mol)

表1 B3PW91/6-311++g(d,p)水平上所有反应物、过渡态和产物的振动频率
通道 3中,H原子首先进攻C2F6中的C-C键,使得两个相连的C原子分开,而H原子与其中一个C原子形成新的C-H键,最终形成裂解产物P2(CF3+CF3H)。该反应所需经过的过渡态TS3的虚频值为-1 008.2 cm-1。从反应物到TS3,C2F6中C-C键长由原来的0.155 4 nm拉长为0.178 8 nm,同时C-H键之间的距离缩短到0.134 8 nm。产物P2的能量为-58.8 kJ/mol,该反应同样为放热反应。TS3的能垒高度为223.5 kJ/mol,比通道 2高出92.9 kJ/mol。因此,比较三条反应通道可知,通道 2为标题反应的主反应通道。
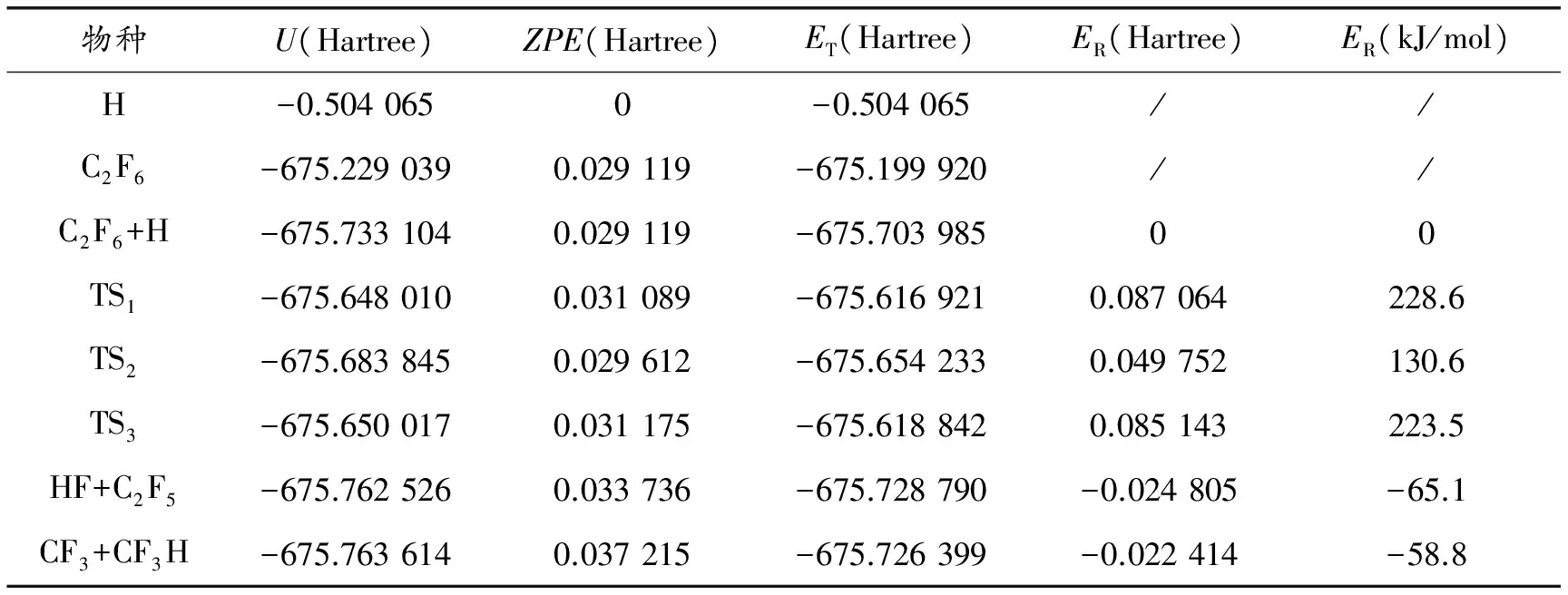
表2 各物种的电子结构能(U)、零点能(ZPE)及总能量(ET)和相对能量(ER)
2.2 反应速率常数
用传统过渡态理论(TST)计算3条反应通道在200 ~2 000 K温度下的速率常数,公式如下:
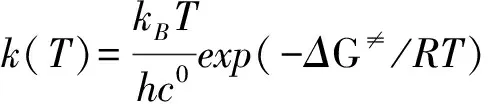
其中:kB是Boltzmann常数(1.380 64910-23JK-1),h是Planck常数(6.626 069 610-34Js),c0是标准校正(2.687 191019moleculescm-3),R是摩尔气体常数,ΔG≠是考虑了零点能校正后的决速步活化吉布斯自由能。表3列出了200~2 000K温度范围内3条反应的速率常数k1、k2和k3随温度变化情况。由表3可知:(1) 在同一温度下,k2的值总是小于k1和k3,验证了上文中势能面的分析结果,即通道 2具有最低的反应能垒;k1和k3的数值比较接近,主要是由于通道 1和 通道 3具有相近的势垒。(2) k1、k2和k3的数值随着温度的升高均出现增大趋势,为正温度系数效应。
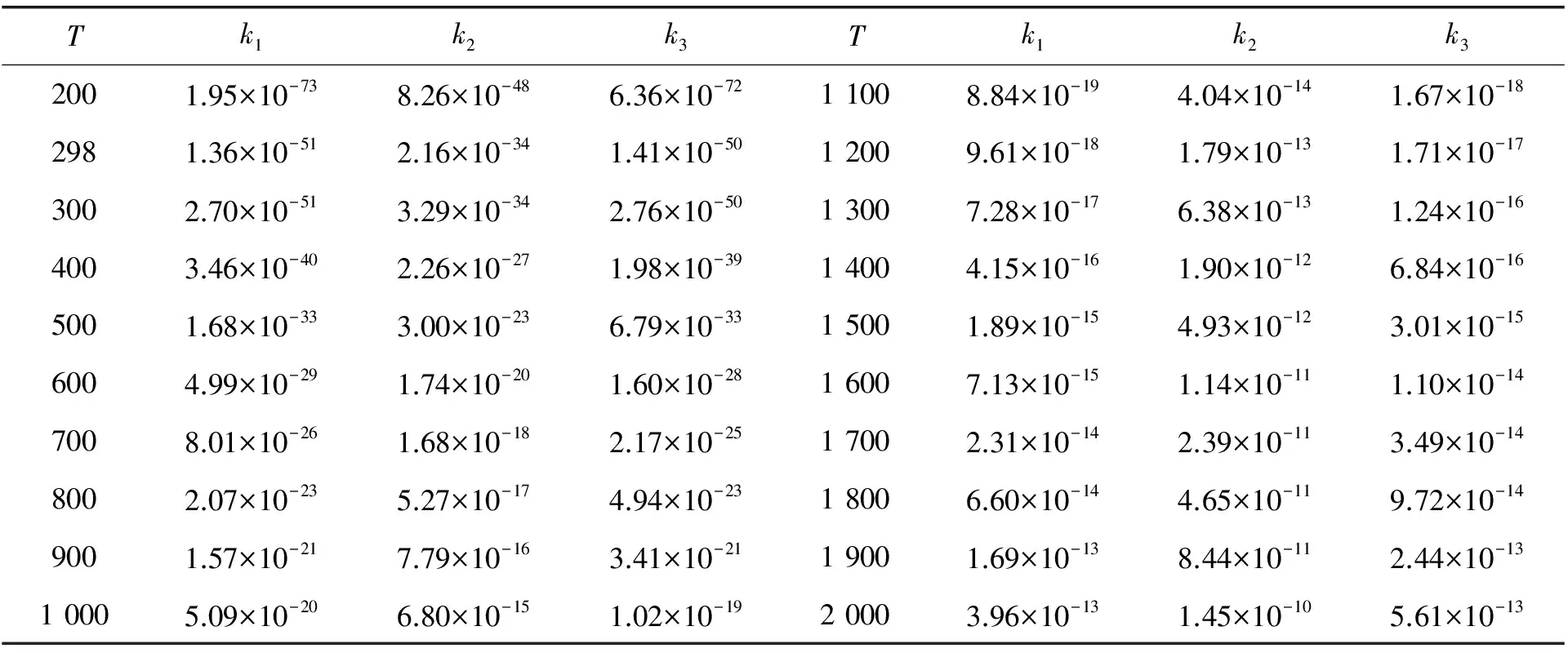
表3 200 K至2 000 K温度范围内各反应通道的速率常数k(cm3·molecule-1·s-1 )随温度的变化
3 结语
用B3PW91/6-311++g(d,p)计算方法研究了C2F6与H自由基的反应机理,获得了3条反应通道。结果表明:(1) 通道 2:C2F6+ H→TS2→P1(C2F5+HF)需要克服的能垒为130.6 kJ/mol,是所有通道势垒中最低的,为该反应体系的主要通道,产物P1为主产物;(2) 3条反应通道的速率常数均随温度的升高而增大,呈正温度系数效应。
[1] 何林,韩军,王光辉,等.六氟乙烷的热分解特性[J].高等学校化学学报,2009,(1):125-128.
[2] 杨健芳.六氟乙烷(FC-116)应用前景和市场分析[J].浙江化工,2008,(10):14-17.
[3] 徐寿昌.有机化学[M].第2版.北京:高等教育出版社,1933.
[4] C.Atterbury,R.A.Kennedy,C.A.Maybew.A study of the reaction of trifluoromethyl sulfur pentafluo- ride,SF5CF3,with several positive ions of atmospheric interest[J].Phys.Chem.Chem.Phys,2001,(3):1949-1953.
[5] C.Atterbury,A.D.J.Critchley,R.A.Kennedy.A study of the gas phase react ions of various cations with two derivatives of SF6,SF5CF3and SF5Cl[J].Phys.Chem.Chem.Phys,2002,(4):2206-2223.
[6] 张仁熙,沈燕,黄丽,等.新温室气体SF5CF3与还原性自由基的反应研究[J].化学世界,2007,(增刊):195-196.
[7] 姚焕英,李林优,侯雯雯,等.SF5CF3与H反应机理的理论研究[J].渭南师范学院学报,2014,29(23):12-23.
[8] 刘艳,任宏江.SF5CF3与还原性自由基C2H3脱氟反应机理研究[J].分子科学学报,2015,(5):368-372.
[9] A.D.Becke.Density-functional thermochemistry.III.The role of exact exchange[J].J.Chem.Phys,1993,(98):5648-5652.
[10] K.P Burke,J.P.Y.Wang,J F.Dobson,et al.Electronic density functional theory:recent progress and new directions[M].New York:Plenum Press,1998.
[11] C.Gonzalez,H.B.Schlegel.An improved algorithm for reaction path following[J].J.Chem.Phys,1989,(90):2154-2162.
[12] C.Gonzalez,H.B.Schlegel.Reaction path following in mass-weighted internal coordinates[J].J.Phys.Chem,1990,(94):5523-5527.
[13] M.J.Frisch,G.W.Trucks,H.B.Schlegel,et al.Gaussian 09[Z].Revision A.02.Wallingford CT:Gaussian Inc,2009.
[14] M.W.Chase,C.A.Davies,R.J.Downey,et al.JANAF thermochemical tables[J].J.Phys,Chem Ref Data,1985,(14):655.
【责任编辑 马小侠】
Theoretical Study on the Reaction Mechanism of C2F6with H Radical
LIU Yan1,2,YU Hong1
(1.School of Chemistry and Materials,Weinan Normal University,Weinan 714099,China; 2.School of Chemistry and Chemical Engineering,Shaanxi Normal University,Xi’an 710062,China)
The density function method B3PW91/6-311++g (d,p) was employed to study the reaction mechanism of C2F6and H radical.Optimized geometries of reactants,transition states and products were verified by the analysis of vibration frequency and intrinsic reaction coordinate (IRC).The results indicate that three possible reaction paths are obtained.The path C2F6+H→TS2→C2F5+HF is the main reaction and product C2F5+HF is the main products by the lowest energy barrier of 130.6 kJ/mol.
C2F6; H radical; reaction mechanism
O643.12
A
1009-5128(2017)08-0047-05
2016-10-10
国家自然科学基金项目:全氟类强温室气体生成及降解机理的理论研究(21503150);渭南师范学院人才基金项目:渭南大气及气候变化的机理研究(15ZRRC07);渭南师范学院特色学科建设项目:秦东化工、材料技术调查(14TSXK04)
刘艳(1978—),女,陕西渭南人,渭南师范学院化学与材料学院副教授,工学博士,主要从事量子化学与计算研究。
