基于多标签直推学习的抗菌肽及其抗菌功能预测
2017-06-01布晓婷,曹隽喆,顾宏
布 晓 婷, 曹 隽 喆, 顾 宏
( 大连理工大学 控制科学与工程学院, 辽宁 大连 116024 )
基于多标签直推学习的抗菌肽及其抗菌功能预测
布 晓 婷, 曹 隽 喆, 顾 宏*
( 大连理工大学 控制科学与工程学院, 辽宁 大连 116024 )
抗菌肽是广泛存在于生物体内的一类具有广谱抗菌作用的天然多肽,因其不易导致细菌耐药性,已成为医药界开发新型抗菌制剂的主要选择,识别出更多的抗菌肽并预测其抗菌功能具有重要意义.提出了一种基于多标签直推学习的抗菌肽及其抗菌功能的预测方法,该方法利用K-spaced氨基酸对组成方法提取多肽特征,采用多标签学习框架和加权近邻图构建直推预测模型,通过对有标签训练样本和无标签待测样本的共同学习来提升预测性能.该方法不仅能够识别多肽是否为抗菌肽,还能同时预测出抗菌肽所具有的单种或多种抗菌功能,且适用于对多效抗菌肽和普通抗菌肽的预测.数值实验表明,与已有的iAMP-2L预测方法相比,所提方法在全局预测精度和多标签预测性能上均有较大提升.
抗菌肽;多标签学习;直推学习;K-spaced氨基酸对组成方法
0 引 言
抗菌肽(antimicrobial peptide,AMP)是广泛存在于生物体内的具有抗菌活性的多肽,一般由5~100个氨基酸构成,是生物体先天免疫系统的重要组成部分.抗菌肽具有广谱抗菌性,对真菌、原虫、病毒及癌细胞等都具有强效的杀伤作用,并且耐药性致病菌也不易对其产生抗药作用[1].目前抗生素滥用问题日益严重,医药界对于新型抗菌药物的需求愈加强烈,近年来不少学者都致力于利用抗菌肽开发新型抗菌剂[2].但由于抗菌肽抗菌功能类别多样,抗菌作用机理复杂,业界对各类抗菌肽的结构和抗菌作用机理都不甚了解,发现更多的抗菌肽并了解其抗菌功能是解决这一问题的有效途径.然而现阶段已发现的抗菌肽种类不多,还有相当大数量的天然抗菌肽未被识别,甚至对于已知抗菌肽的具体抗菌功能了解得也不够全面.
针对上述问题,研究人员主要采用基于实验的方法和基于计算的方法来对抗菌肽识别加以解决.实验方法是通过实验分离测定的方式对多肽的抗菌活性进行观察判定,该方法的优点是识别精度高,但操作过程复杂,需要投入大量的物力、人力及时间成本[3],效率较低且不具备大规模操作性,随着医药界对抗菌肽新型制剂开发的不断深入,这种方法越来越难以满足研究需求.而随着生物信息学的迅猛发展,基于计算的方法被应用于该领域,其中基于机器学习的计算方法以其高精度、低成本、高可行性及高可靠性等优势,被越来越多地应用于抗菌肽及其抗菌功能的预测中.机器学习方法通过对大量抗菌肽生物数据的分析和学习,不仅能够发现抗菌功能同生化属性之间的线性关系,还可以挖掘内在的非线性关联,这对于深入挖掘大规模无序数据中隐藏的生物信息,更深入地了解抗菌肽的分子组成结构、基因表达机制及抗微生物作用机理有十分积极的作用.
基于机器学习的抗菌肽预测属于分类学习问题,主要的步骤为建立数据集、提取多肽特征、设计分类器.最初发现的抗菌肽只具有一种抗菌活性,因此早期的研究主要用于推断待测多肽是否为抗菌肽,即二分类问题,常见的算法包括神经网络[4-5]、支持向量机[6-7]和随机森林[8-9]等.而随着越来越多的多效抗菌肽(即同时具有多种抗菌功能的抗菌肽)被发现,近年来一些学者开始考虑对抗菌肽的多效抗菌功能进行预测,如Joseph等[10]提出了基于随机森林和支持向量机的多标签预测方法ClassAMP,用以区分抗细菌肽、抗真菌肽和抗病毒肽3类不同功能的抗菌肽;Zou等[11]提出了基于序列信息和多标签学习的LIFT预测方法,用以预测抗菌肽是否具有抗细菌、抗病毒、抗真菌等8种不同抗菌活性.这两种方法只是针对确定为抗菌肽的样本进行功能预测,而不能判断一条多肽是否为抗菌肽.而Xiao等[12]提出的iAMP-2L预测方法则同时考虑了对抗菌肽预测和对其功能的预测,该方法分为两个阶段,先利用二分类算法鉴别某多肽是否为抗菌肽,然后对被鉴别为抗菌肽的多肽采用多标签学习算法进行抗菌功能的预测.
总的来说,目前能对多效抗菌肽预测的方法非常少,现有的方法都是采用多标签学习算法进行处理.然而多标签学习的初衷是为了解决歧义性问题,样本通常都是具有一个或多个正标签的正类样本,一般不存在负类样本,而抗菌肽的预测问题则完全不同,一条多肽完全可以是不具有任何抗菌活性的非抗菌肽,其本质是个具有负类样本的预测问题,这是传统的多标签学习所无法直接处理的.因此,文献[10]和[11]的方法为了避开这个问题,只针对抗菌肽进行功能预测.而在实际应用中,待测样本的属性其实是完全未知的,完整的预测任务应该包含两部分:首先判断出这些样本是否为抗菌肽,然后再对确定为抗菌肽的样本进一步预测其抗菌功能.而文献[12]的方法则采用这两部分先后处理的方式,这种两阶段鉴定方法不仅过程复杂,并且两个阶段的误差叠加导致预测精度不高,而且还割裂了两个预测问题的联系.
针对上述问题,本文提出一种新的基于多标签直推学习的抗菌肽及其抗菌功能预测方法,该方法将预测问题看作一个可以含有负标签样本的特殊多标签学习问题来处理,不仅具有预测多效抗菌肽的能力,还能在一个学习算法下将抗菌肽预测及其抗菌功能预测两个任务同时完成.
1 实验数据
本文建立了两个数据集:基准数据集S1和独立测试集S2,其中基准数据集S1用于训练和交叉验证,独立测试集S2用于验证预测方法的泛化性.S1中的数据来自文献[12],并对其中的个别错误进行修正得到,S2中的数据由文献[12]的独立测试集部分提取得到.两个数据集均包括抗菌肽数据和非抗菌肽数据.
基准数据集S1包括两大类:抗菌肽数据和非抗菌肽数据,其表示方法如下:
(1)
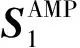

表1 基准数据集S1各类别数量
为了更好地验证本预测方法的泛化性,本文还使用独立测试集S2进行独立测试集实验,其中无重复统计的抗菌肽数量为350.独立测试集S2各类别数量见表2.

表2 独立测试集S2各类别数量
2 原理和方法
2.1 多肽序列特征信息的提取
对于某一多肽而言,其肽链的结构直接决定着其生物学功能如抗菌功能,而一级结构作为最基本的结构直接决定着其二级结构和三级结构,故多肽的一级结构对于其抗菌功能的有无及具体类型至关重要.多肽的一级结构可以由组成它的氨基酸序列表示,本文特征信息提取的出发点即将多肽的氨基酸序列信息转化为可量化的特征向量,通过特征向量尽可能形象地将特定多肽表达出来.
本文使用K-spaced氨基酸对组成方法(composition ofK-spaced amino acid pairs,CKSAAP) 对样本进行特征提取.CKSAAP首先由Chen等[13]于2007年提出并用于蛋白质灵活化区域预测中,该方法的优点在于充分利用蛋白质或多肽序列中各个氨基酸的局部相互作用信息,故在不少生物信息学领域都获得了不错的实验结果,例如蛋白质磷酸化作用位点预测[14]、三型效应蛋白鉴别[15]、赖氨酸甲基化位点及甲基化度预测[16]等领域.由于抗菌肽的氨基酸序列通常较短,最短的抗菌肽仅由5个氨基酸构成,而CKSAAP 作为一种关注于组成多肽的各个氨基酸局部相互作用信息的特征提取方法,对短肽特征的刻画较为出色.通过前期比较发现,相对于关注蛋白质长链氨基酸出现概率的氨基酸组成方法(amino acid composition,AAC)和关注氨基酸出现概率和物化性质的伪氨基酸组成方法(pseudo-amino acid composition,PseAAC),CKSAAP 对本文所研究问题的表征效果更好一些,因此本文选定CKSAAP提取的特征用于最终的预测.当K=0时,该方法将多肽一级结构所蕴含的信息提取为以下向量:
P=(NAANAC…NAV)T
(2)
其中NXY表示序列中氨基酸X与氨基酸Y连续出现的次数,X与Y可以是相同的氨基酸,由于组成多肽的基本氨基酸为20个,氨基酸两两任意组合会产生20×20种可能,故该情况下特征向量维数为400.当K=1时,特征向量为
P=(NAANAC…NAVNAxANAxC…NAxV)T
(3)
其中前400行与式(2)相同,401至800行中NXxY表示序列中氨基酸X与氨基酸Y中间相隔一个氨基酸的情况出现的次数,其中x表示任意一个氨基酸.K=2、3、4时对应的1 200、1 600、2 000 维特征向量依此类推.在本文实验中,选择K=0即特征维度最小的情况,此时每个多肽均由一个400维的特征向量表示.
2.2 基于多标签直推学习的预测方法
直推学习(transductive learning)由Vapnik[17]于1995年首次提出,现已应用于文本识别[18]、视觉跟踪[19]、蛋白质亚细胞定位[20]等多个领域并取得了不错的效果.该方法不同于传统的归纳演绎式学习方法,在构建方法的过程中除了使用训练集中的信息之外,将待测试样本中的信息也利用起来进行方法构建.通常这种学习方法应用在无标签样本数量较大而有标签样本数量不够多的问题中,将测试集合信息也用于预测方法的构建能够使预测方法更好地识别整个空间的数据特性[21],从而使预测方法具有更好的预测性能.
对于本文的研究问题而言,目前已知的抗菌肽非常有限,还有数量十分大的抗菌肽未被发现,传统的预测方法仅使用已知抗菌肽的信息来构建预测模型而忽视了未知测试集中包含的大量信息,往往不能得出准确率较高的预测结果,而直推学习方法恰恰能够使有效信息得以利用从而得到不错的预测结果.本文在利用直推学习方法构建近邻图时,在对各样本局部关联关系计算时对各抗菌功能类别加以不同权重,将不同类别对预测方法的贡献度区分开来,从而使得基于多标签直推学习的抗菌肽及其抗菌功能预测方法预测结果更佳.本文构建的预测模型如图1所示.


图1 基于多标签直推学习的抗菌肽及其抗菌功能预测模型示意图
Fig.1 Diagram of the model for predicting the antimicrobial peptides and their functional types based on multi-label transductive learning

(4)
其中N(Xi)表示样本Xi的近邻集合;S(i,j)表示样本Xi与样本Xj在特征空间的相似度;C(i,j)表示样本Xi与样本Xj在标签空间的相似度.显然,对于矩阵W,其任意行向量的元素之和均为1.
采用高斯核(Gaussian kernel)函数计算特征空间的相似度S(i,j),特征空间越相似则值越大,其具体定义如下:
(5)
其中d表示距离度量,本文选用欧氏距离;μ为超参数,本文取为2;σ为调节参数,本文取所有样本之间的平均距离.
C(i,j)表示样本Xi与样本Xj在标签空间的相似度,标签空间越相似则值越大.当样本Xi和Xj不都是待测样本时,有
(6)

(7)
其中Np表示训练集中抗菌肽的样本个数,Nt表示训练集中具有第t个标签的样本个数.这种权重确定方法减弱了那些在大多数样本中存在的标签的重要程度,同时增强了一些在小部分样本中出现的低频标签的重要程度,即若训练集中具有第t个标签的样本个数越少,则该标签的权值越大,可以认为第t个标签具有较好的类别区分能力,对多标签分类有较大帮助[22].由表1可以看出,具有抗细菌标签的抗菌肽个数为770,而具有抗HIV标签的抗菌肽个数为85,显然这两种抗菌功能标签对于分类的贡献度不同,即具有一定的不平衡性,因此将其赋予依据数量确定的不同的权值是有必要的.
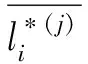
在最优化确定信任度之前,需将训练集标签向量进行预处理.对于训练集中的抗菌肽样本,有
(8)
依据上文的平滑性假设本文提出一个求解信任度的最优化问题:

(9)
最优化目标是最小化相似样本的标签之间的加权差,为了简化上式,有以下公式:
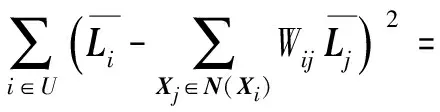
(10)
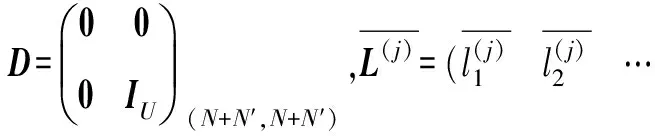
这样式(9)便能简化成下式:
(11)
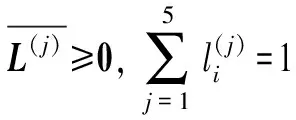
为求解待测样本Xi所含标签个数θi,本文提出一个最优化问题:

(12)
式(11)和(12)表示的优化问题均有唯一最优解,文献[23]对此给出了理论证明,因此可求得直推的结果.预测算法流程如下:
输入:
预测算法:
构建有限加权近邻图,并确定近邻间边的权重矩阵W,见式(4);

输出:
2.3 评价指标
本文研究的课题属于一种特殊的多标签问题,能够同时将正负类样本和正类样本的一个或多个抗菌功能标签预测出来,故本文将使用全局评价指标和多标签评价指标这两类指标来衡量预测方法的效果.
全局评价指标[24]为二分类指标,主要用来评价预测方法对于抗菌肽和非抗菌肽的分类效果,若某待测样本的预测标签全为0则被预测为非抗菌肽,否则是抗菌肽.全局评价指标包括敏感性(sensitivity,Ssn)、特异性(specificity,Ssp)、正确率(accuracy,A)和马氏相关系数(Mathew′s correlation coefficient,Mc),其具体公式如下:
(13)
(14)
其中Tp(true positive)表示抗菌肽被预测为抗菌肽的个数,Tn(true negative)表示非抗菌肽被预测为非抗菌肽的个数,Fp(false positive)表示非抗菌肽被预测为抗菌肽的个数,Fn(false negative) 表示抗菌肽被预测为非抗菌肽的个数,如式(14),N表示符合某条件的多肽的个数,其中N的上角标表示某多肽实际为抗菌肽或非抗菌肽,分别用+和-两符号表示,N的下角标表示某多肽被预测为抗菌肽或非抗菌肽,表示方法同上.对于以上4个全局评价指标,Ssn、Ssp和A的取值范围均为[0,1],Mc的取值范围为[-1,1],并且它们的值越大则表示预测方法越好.
多标签评价指标[25]主要用来评价预测方法对于抗菌肽样本所含标签的预测准确度,它包括汉明损失(Hamming Loss,Hl)、准确度(accuracy,Ac)、查准率(precision,P)、查全率(recall,R)和完全正确率(absolute true,At),其具体公式如下:
(15)

(16)
以上这5个多标签评价指标的取值均为[0,1],准确度、查准率、查全率和完全正确率均越大越好,而汉明损失越小越好.
3 实验结果
本文在构建近邻图时首先需确定该方法的参数即近邻数K,为避免结果的随机不确定性,本文选用留一法(leave-one-out,LOO).为了获得预测性能最佳的预测方法,以基准数据集S1上的全局指标中的正确率指标最大来选取最优的近邻数K,本文对K取1至30均进行了实验,实验结果表明,当K为3时,S1上的全局指标中的正确率最大,由此确定K=3.表3选列了部分K下的全局指标中的正确率指标结果统计表.


表3 不同近邻数K下的正确率结果

表4 S1上的全局评价指标结果
本文预测方法的所有指标结果均优于iAMP-2L预测方法,其中全局指标中的准确率高达98.20%,多标签评价指标中的汉明损失也仅为0.110 1.实验结果表明,本文预测方法的全局性能和多标签学习性能比现有方法有了大幅提升.

表上的多标签评价指标结果

表6 S2上的全局评价指标结果
表上的多标签评价指标结果
由以上两表看出,本文预测方法在独立测试集S2上的预测表现依然很好,全局评价指标中所有指标都优于iAMP-2L预测方法,其中正确率指标高达98.43%,而多标签评价方面指标结果虽然比在基准数据集S1上略差,但汉明损失也仅有0.122 9.总的来说,本文预测方法比现有方法具有更好的泛化性能.
对于多标签评价问题,除了从以上指标分析方法性能外,本文还对不同标签数的样本预测完全正确率进行了统计,结果见表8.从表8可以看出,本文预测方法对于样本标签数为1、2、3、4的样本的预测完全正确率均高于iAMP-2L预测方法,其中前3个预测完全正确率都超过了45%.而对于同时具有5个标签的样本来讲,本文预测方法效果相对欠佳,主要原因在于本文的一体化预测方法是在有负样本参与的情况下进行的,其直推学习算法采用了样本的部分聚类信息进行学习,一些边缘负样本对标签的推断具有比较大的影响,尤其是对具有多个正标签的样本影响更加明显,而iAMP-2L预测方法在第2阶段中提前把负样本剔除,因此影响较小.另外为了能够生成负样本分类结果,算法在每类标签上的学习都是相对独立的,而对标签间的关联性学习不够,也影响了对标签的学习效果,这一点在后续的研究中需要改进.

表上的不同标签数预测完全正确率
4 结 语
本文构建的基于多标签直推学习的抗菌肽及其抗菌功能预测方法能够一次性进行抗菌肽的鉴别及其抗菌功能的鉴定工作,该方法在构建近邻图时将各类别标签加以不同权重,将不同标签对预测方法的贡献度区分开来,并且突破传统的利用样本特征信息计算样本局部关联关系的方法,将样本的标签信息加入到局部关联关系的公式中,使计算出的各近邻样本的关联关系更贴近真实值.本文预测方法将直推学习算法应用在抗菌肽预测领域,充分适应了抗菌肽领域未知抗菌肽数量远远大于已知抗菌肽数量以及抗菌肽序列间同源性较低的特点,有效提高了对待测样本抗菌功能的预测精确度.为了将算法实际应用于抗菌肽的实验判定,下一步计划基于本文预测方法开发抗菌肽在线预测平台,为相关研究人员提供高精度在线预测服务.
[1] ZASLOFF M. Antimicrobial peptides of multicellular organisms [J]. Nature, 2002, 415(6870):389-395.
[2] HAMMAMI R, FLISS I. Current trends in antimicrobial agent research: chemo- and bioinformatics approaches [J]. Drug Discovery Today, 2010, 15(13/14):540-546.
[3] KHOSRAVIAN M, FARAMARZI F K, BEIGI M M,etal. Predicting antibacterial peptides by the concept of Chou′s pseudo-amino acid composition and machine learning methods [J]. Protein and Peptide Letters, 2013, 20(2):180-186.
[4] TORRENT M, ANDREU D, NOGUÉS V M,etal. Connecting peptide physicochemical and antimicrobial properties by a rational prediction model [J]. PLoS One, 2011, 6(2):e16968.
[5] HOLTON T A, POLLASTRI G, SHIELDS D C,etal. CPPpred: prediction of cell penetrating peptides [J]. Bioinformatics, 2013, 29(23):3094-3096.
[6] VIJAYAKUMAR S, PTV L. ACPP: A web server for prediction and design of anti-cancer peptides [J]. International Journal of Peptide Research and Therapeutics, 2015, 21(1):99-106.
[8] CHANG K Y, YANG J R. Analysis and prediction of highly effective antiviral peptides based on random forests [J]. PLoS One, 2013, 8(8):e70166.
[9] KARNIK S, PRASAD A, DIWEVEDI A,etal. Identification of defensins employing recurrence quantification analysis and random forest classifiers [J]. Lecture Notes in Computer Science, 2009, 5909: 152-157.
[10] JOSEPH S, KARNIK S, NILAWE P,etal. ClassAMP: a prediction tool for classification of antimicrobial peptides [J]. IEEE-ACM Transactions on Computational Biology and Bioinformatics, 2012, 9(5):1535-1538.
[11] ZOU Hongliang, XIAO Xuan. A new multi-label classifier in identifying the functional types of human membrane proteins [J]. The Journal of Membrane Biology, 2015, 248(2):179-186.
[12] XIAO Xuan, WANG Pu, LIN Weizhong,etal. iAMP-2L: A two-level multi-label classifier for identifying antimicrobial peptides and their functional types [J]. Analytical Biochemistry, 2013, 436(2):168-177.
[13] CHEN Ke, KURGAN L A, RUAN Jishou. Prediction of flexible/rigid regions from protein sequences usingk-spaced amino acid pairs [J]. BMC Structural Biology, 2007, 7(1):25.
[14] ZHAO Xiaowei, ZHANG Wenyi, XU Xin,etal. Prediction of protein phosphorylation sites by using the composition ofk-spaced amino acid pairs [J]. PLoS One, 2012, 7(10):e46302.
[15] DONG Xiaobao, ZHANG Yongjun, ZHANG Ziding. Using weakly conserved motifs hidden in secretion signals to identify type-Ⅲ effectors from bacterial pathogen genomes [J]. PLoS One, 2013, 8(2):e56632.
[16] JU Zhe, CAO Junzhe, GU Hong. iLM-2L: A two-level predictor for identifying protein lysine methylation sites and their methylation degrees by incorporatingk-gap amino acid pairs into Chou′s general PseAAC [J]. Journal of Theoretical Biology, 2015, 385(8):50-57.
[17] VAPNIK V. The Nature of Statistical Learning Theory [M]. Berlin: Springer, 1995.
[18] JOACHIMS T. Transductive inference for text classification using support vector machines [C] // Sixteenth International Conference on Machine Learning. Burlington: Morgan Kaufmann Publishers Inc., 1999: 200-209.
[19] ZHA Yufei, YANG Yuan, BI Duyan. Graph-based transductive learning for robust visual tracking [J]. Pattern Recognition, 2010, 43(1):187-196.
[20] CAO Junzhe, LIU Wenqi, HE Jianjun,etal. Identifying the singleplex and multiplex proteins based on transductive learning for protein subcellular localization prediction [J]. Biotechnology Letters, 2013, 35(7):1107-1113.
[21] 陈毅松,汪国平,董士海. 基于支持向量机的渐进直推式分类学习算法[J]. 软件学报, 2003, 14(3):451-460.
CHEN Yisong, WANG Guoping, DONG Shihai. A progressive transductive inference algorithm based on support vector machine [J]. Journal of Software, 2003, 14(3):451-460. (in Chinese)
[22] 蒋 健. 文本分类中特征提取和特征加权方法研究[D]. 重庆: 重庆大学, 2010.
JIANG Jian. Study on feature selection and feature weighting of text classification [D]. Chongqing: Chongqing University, 2010. (in Chinese)
[23] KONG Xiangnan, NG M K, ZHOU Zhihua. Transductive multilabel learning via label set propagation [J]. IEEE Transactions on Knowledge and Data Engineering, 2013, 25(3):704-719.
[24] CHEN Wei, FENG Pengmian, LIN Hao,etal. iRSpot-PseDNC: identify recombination spots with pseudo dinucleotide composition [J]. Nucleic Acids Research, 2013, 41(6):e68.
[25] MAIMON O, ROKACH L. Data Mining and Knowledge Discovery Handbook [M]. Heidelberg: Springer, 2010.
Prediction of antimicrobial peptides and their functional types based on multi-label transductive learning
BU Xiaoting, CAO Junzhe, GU Hong*
( School of Control Science and Engineering, Dalian University of Technology, Dalian 116024, China )
Antimicrobial peptides, a type of natural polypeptides with broad-spectrum antimicrobial activity, are widely found in organisms. Because of a slim chance of bacterial resistance, antimicrobial peptides have become a preferred option for the pharmaceutical industry to develop new antibacterial preparations. In this sense, it is of great significance to identify more antimicrobial peptides and then make clear their antimicrobial functional types. In view of this fact, a prediction method based on multi-label transductive learning is proposed to predict antimicrobial peptides and their functional types. This method extracts the polypeptide characteristics by composition ofK-spaced amino acid pairs and constructs transductive prediction models by the weighted neighbor graph and multi-label learning framework. Through the study of labeled training data and unlabeled data to be tested, this method can not only predict whether a polypeptide is an antimicrobial peptide, but also predict what type of antimicrobial function a polypeptide would have. In addition, this method is applicable to both multiple-effect antimicrobial peptides and common antimicrobial peptides. Numerical experiments have shown that the proposed method is more accurate than iAMP-2L method in performance in terms of overall prediction and multi-label prediction.
antimicrobial peptides; multi-label learning; transductive learning; composition ofK-spaced amino acid pairs (CKSAAP)
1000-8608(2017)03-0293-09
2016-09-20;
2017-03-23.
国家自然科学基金资助项目(U1560102,61502074);中国博士后科学基金资助项目(2016M591430);大连理工大学基本科研业务费资助项目(DUT15RC(3)030).
布晓婷(1991-),女,硕士生,E-mail:pudding_bxt@126.com;曹隽喆(1984-),男,讲师;顾 宏*(1961-),男,教授,博士生导师,E-mail:guhong@dlut.edu.cn.
TP181
A
10.7511/dllgxb201703012
