主要果树植物全基因组测序研究进展
2017-04-05张俊环孙浩元杨丽姜凤超王
张俊环++孙浩元++杨丽++姜凤超++王玉柱

摘要:近几年,果树植物全基因组测序研究迅速升温,多个果树基因组图谱被陆续公布,为果树分子生物学和比较基因组学研究提供了大量的数据信息。通过比较分析已经完成全基因组测序的11种我国主栽果树的测序研究结果,就果树植物的起源和进化、重要农艺性状相关基因的发掘以及测序结果的应用前景方面进行了概述。
关键词:果树;全基因组;测序;进化;功能基因
中图分类号:Q78文献标志码:A[HK]
文章编号:1002-1302(2016)12-0006-06[HS)][HT9.SS]
收稿日期:2016-07-27
基金项目:国家自然科学基金(编号:31270709、31401836);北京市自然科学基金(编号6162012)。
作者简介:张俊环(1974—),女,山东菏泽人,博士,副研究员,现主要从事果树分子生物学研究工作。Tel:(010)82595857;E-mail:zhang_junhuan@163.com。
通信作者:王玉柱,博士,研究员,现主要从事果树育种研究工作。Tel:(010)82592521;E-mail:chinabjwyz@126.com。
果树作为重要的经济作物,在国内外农业生产中均占有重要的地位。但是由于果树生命周期较长、基因组的杂合度较高、重复序列较多、且大多果树因自交不亲和而导致遗传背景不清晰,这些因素限制了果树分子生物学研究和全基因组测序研究的进程。然而,近些年,随着测序技术的发展、测序效率的提高和测序成本的降低,果树植物的全基因组测序工作在全球迅速展开,自2007年完成第一个果树植物葡萄(Vitis vinifera)基因组测序以来,不到10年时间,已有14种果树植物的全基因组测序工作相继完成。这些果树种类的全基因组测序结果为果树分子生物学研究搭建出了庞大的资源平台,不仅有助于了解果树的基因组结构和功能,而且对于探索果树植物的起源与进化、开展重要功能基因的定位和克隆、加速分子育种进程等均具有重要的指导意义。本文通过分析其中11种我国主栽果树的全基因组测序研究结果,围绕果树植物尤其是蔷薇科植物的起源和进化、典型功能组分的代谢通路及其相关基因,以及测序结果的应用前景方面进行分析和讨论。
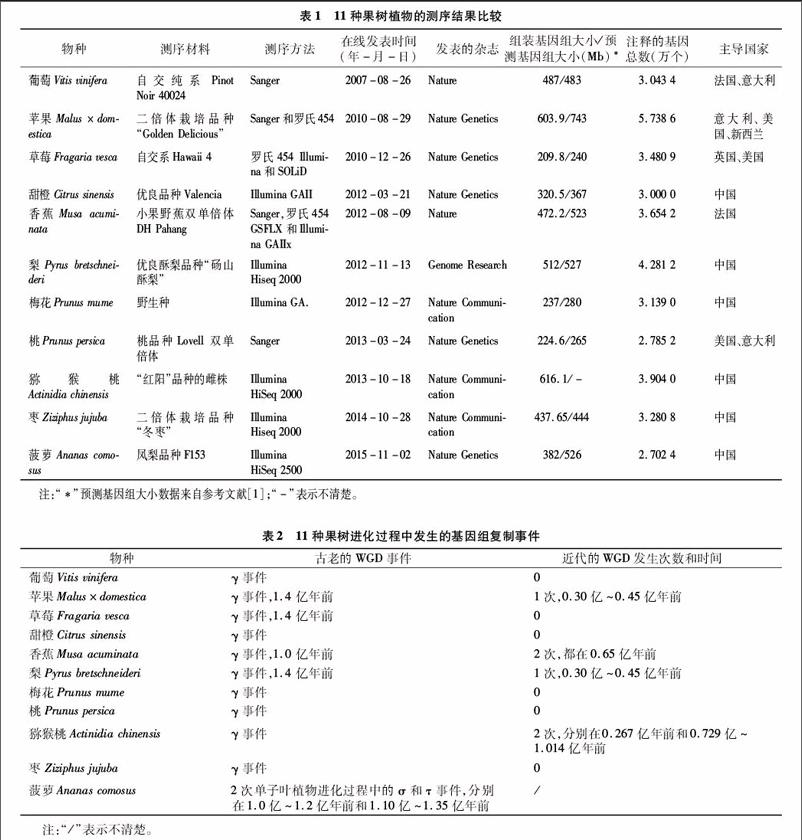
1主要果树植物的测序结果基本数据
已完成全基因组测序并公布草图的14种果树植物中,既包括热带亚热带常绿果树香蕉、甜橙、番木瓜和菠萝,也包括北方落叶果树苹果、梨、枣等,其中我国自主完成测序的树种就有6个。测序材料除甜橙(Citrus sinensis)采用纯合度相对较高的双单倍体材料外,其余5个树种均是采用遗传背景相对不清晰、杂合度较高的二倍体栽培品种(梨Pyrus bretschneideri、枣Ziziphus jujuba、猕猴桃Actinidia chinensis和菠蘿Ananas comosus)或野生品种(梅花Prunus mume)(表1)[1]。测序技术主要采用第二代测序技术Illumina平台。与第一代Sanger测序技术相比,第二代测序技术降低了测序成本,提高了测序速率,且测序覆盖度更高,尤其是Illumina HiSeq 2000测序技术平台以高通量、高分辨率、高精度和价格低廉等优势发挥着巨大作用,已成功应用于多种果树植物的全基因组或转录组测序研究。
2果树植物的起源和进化事件
已完成测序的植物基因组进化过程研究结果表明,全基因组复制事件几乎发生于每个植物的进化过程中。全基因组复制存在古老的全基因组复制(old whole-genome duplications,old WGD)和近代的全基因组复制(recent whole-genome duplications,recent WGD)2种方式。在双子叶植物中,古老的全基因组复制也被称为古六倍体化(paleohexaploidization)进化或者三倍化复制(triplicated arrangement),又称γ事件[2-7]。单子叶植物中古老的全基因组复制事件与双子叶植物有所不同,包括ρ、σ和τ等3种不同的进化事件[8]。近代的全基因组复制过程伴随基因的丢失和基因新功能的产生,是大多数双子叶植物进化的主要动力。表2给出了这11种果树植物在进化过程中所发生的全基因组复制事件。
由表2看出,这11种果树植物全部都经历了古老的WGD,并且菠萝还发生了2次。已完成测序的这些真双子叶植物基因组的六倍体化过程都发生在相似的时间,即是在单子叶植物与双子叶植物的分化之后,蔷薇类分支(Eurosids clade)分化之前,早于葡萄科和蔷薇科的分化,也早于鼠李科(Rhamnaceae)和蔷薇科(Rosaceae)的分化,大约在1.4亿年前。在约0.872亿年前,枣和蔷薇科(包括梨、苹果、桃、梅和草莓)发生了分化[9]。葡萄基因组首次解析了全基因组三倍化复制事件,被视为古六倍体化进化机制的实例,随后苹果、草莓、甜橙等基因组结构的分析均支持双子叶植物祖先的古代六倍体是单一起源的假说。另外,值得注意的是,作为单子叶植物的香蕉基因组,在进化过程中,没有发生禾本科植物的ρ、σ或τ复制,而是发生了与双子叶植物相似的γ复制事件[10]。[FL)]
由表2还可以看出,相对于进化过程中普遍发生的古老的全基因组复制事件,在这11种果树植物中仅有苹果、梨、猕猴桃和香蕉4种果树基因组发生了近代的WGD事件。通过共线性区域分析,发现猕猴桃进化过程中发生3次基因组倍增历史事件,1次古老的全基因组复制(γ事件)和2次近代的全基因组复制事件(Ad-α和Ad-β),后2次分别发生在0.267亿年前和0.729亿~1.014亿年前[6]。蔷薇科的梨与苹果、草莓则都经历了1.4亿年前双子叶植物所共有的古六倍化事件,并发生了1次全基因组复制事件。之后大约在 0.30 亿~0.45亿年前,梨和苹果又经历了1次全基因组复制事件,而梨与苹果的分化大约发生在0.054亿~0.215亿年前[11]。同为蔷薇科的草莓、梅、桃与苹果发生分化后,并没有出现过较近时期的全基因组复制事件[4-5,12],葡萄、甜橙、枣基因组在近代也均未发生全基因组复制事件[2,7,9]。枣的基因组在历史进化中经历了复杂的染色体断裂、融合及片段重组过程。甜橙基因组在从双子叶植物古六倍体祖先进化过程中则发生了频繁的染色体易位和融合事件,至少有49次发生在9条染色体上。特别值得注意的是,在4号染色体上仅发生1次易位和融合事件,而其他染色体均发生多次(3~12次)染色体内的易位和融合过程[7]。
多个蔷薇科果树植物全基因组测序的完成,使人们对蔷薇科植物基因组的起源和进化过程有了更深入的了解。通过草莓属与李属间389个蔷薇科标记在2个基因组间共线性区域的分析,为蔷薇科9条祖先染色体的重建提供了更广泛的证据[4]。在梅全基因组序列图谱的基础上,结合已完成的苹果和草莓基因组序列,分析了它们在进化过程中的染色体变化,进一步成功重建出了蔷薇科植物的9条祖先染色体[5],并深入分析了苹果属、草莓属和李属3个属所分别经历的不同染色体融合、断裂和复制事件,即从9条祖先染色体的进化过程中,梅经历了至少11次的断裂和11次的融合过程,苹果发生了1次近代WGD和5次融合,而草莓则经历了15次的融合过程。梨染色体进化研究再次证明,有9条祖先染色体不仅是苹果亚科的起源,也是整个蔷薇科植物的祖先[11]。
3果树重要经济性状相关基因的揭示
在全基因组测序过程中,每个果树种类都注释到了庞大的基因数目,均在2.5万个以上(表1),通过进一步的分析,发掘出了与抗性增强、果实发育和品质形成相关的重要基因,不仅有利于本物种优质、高抗品种或类型的培育,而且对于其他种类果树抗性及果实品质改良也将起重要借鉴作用。
3.1控制香气物质合成的基因
果实的香气物质主要包括酯类、醇类、酮类、醛类、萜类和挥发性酚类等次生代谢物质,主要来自于萜类代谢、苯丙烷类代谢和脂氧合酶途径[13]。不同果实中香气物质的类型不同,也就形成了不同种类果树的果实有着各自特异的香气。
草莓果实的香味物质主要来源于脂肪酸代谢、萜类化合物代谢和苯丙烷代谢途径,有7个基因家族与这些挥发性组分的产生有关,包括酰基转移酶、萜烯合酶和小分子O-甲基转移酶等[4]。葡萄酒的香气直接与促进萜烯类(树脂、芳香精油类次生代谢物)合成的萜烯合酶(TPSs)基因有关[2]。在葡萄的基因组中,发现有89个与萜烯类合成相关的功能基因和27个拟功能基因。α-亚麻酸代谢途径与梨的香气形成相关。在梨基因组中分析挥发性物质产生的3个主要途径(脂肪酸、氨基酸和碳水化合物)相关的基因,发现在苹果和梨中参与α-亚麻酸代谢途径的脂氧合酶(lipoxygenase,LOX)和乙醇脱氢酶(alcohol dehydrogenase,ADH)的基因数目较多。而促进香味物质释放的β-葡萄糖苷酶基因中,仅有20%在梨果实中表达,大多香味物质仍以结合态存在,这可能是梨香味不明显的原因[11]。Zhang等在对梅花香味分子机制的研究中,首次发现了能直接催化生成梅花花香中重要成分乙酸苯甲酯的苯甲醇乙酰基转移酶BEAT基因,该基因在梅花基因组中显著扩增至34个,远远超过苹果中的16个、草莓14個、葡萄4个。34个基因中有26个是呈簇分布的,最大一簇包括12个基因,并呈串联重复分布。由此推测这些扩张的BEAT基因的剂量效应,增加了乙酸苯甲酯的含量,从而使梅花具有独特的花香[5]。
3.2控制花青素合成的基因
花青素是使果实(包括果皮和果肉)呈现红色的重要成分,是黄酮类化合物的一种,是重要的抗氧化物质,也是果实具有漂亮外观和内在保健价值的重要组分。
Shulaev等对草莓转录因子MYB家族的研究表明,MYB123是草莓花色素合成的重要调节因子。草莓中有187个MYB类转录因子,其中R2R3 MYBs与拟南芥在类黄酮和花青素的合成功能上有较高的同源性。通过拟南芥苯丙烷代谢途径中的20个R2R3 MYBs序列与草莓基因组进行Blast分析,鉴定出25个高度同源的序列,其中扩张最大的分支是控制原花色素水平的MYB123,在草莓中至少有6个成员,是草莓中花色素合成的重要调控因子[4]。已测序的红肉猕猴桃“红阳”果实中也含有较高的花青素,但基因组数据显示猕猴桃中与花青素合成有关的关键酶没有发生扩张,而在类黄酮合成路径中,与葡萄和甜橙相比,猕猴桃中的查尔酮异构酶(chalcone isomerase)、黄烷酮3-脱氢酶(flavanone 3-hydroxylase)和类黄酮3-O-脱氢酶3个基因家族发生了扩张,暗示这3个基因家族与猕猴桃花青素的合成密切相关[6]。
3.3与维生素C积累相关的基因
维生素C,也称L-抗坏血酸(L-ascorbic acid,AsA),其含量是衡量果实营养价值的重要指标之一,特别是枣、猕猴桃和柑橘类均以果实富含维生素C而著称,并得到消费者的广泛喜爱。维生素C积累机制的研究也一直是果树科研工作者努力的方向。
Xu等分析了甜橙果实中AsA上游的4个合成分支途径的关键酶基因,半乳糖醛酸代谢途径的许多基因都发生了上调。特别是半乳糖醛酸酯分支途径的3个关键酶基因(PG、PME和GalUR)发生了明显的上调,其中编码D-半乳糖醛酸还原酶的基因(GalUR)是这一代谢途径的限速酶基因。同时在柑橘基因组中鉴定出18个GalUR同源基因,是已测序植物中最多的。进一步对蔷薇类Malvidae clade家族部内关系较近的4个树种(柑橘、可可、番木瓜和拟南芥)进行进化分析,发现在甜橙内表现扩张的GalUR基因分成2个簇,一簇包括4个基因,另一簇有7个基因,并且可能是串联复制,这2个基因簇在维生素C含量较高的番木瓜中也表现出了扩张。[WTBX][STBX]GalUR-12[WTBZ][STBZ]与草莓中的GalUR基因序列有着高度的相似性和较近的进化关系,并且已经证实该基因与产生特异的维生素C有关。所有证据表明甜橙果实中半乳糖醛酸酯途径的基因发生了高度的扩张,并且GalUR基因在AsA积累过程中发挥最大的作用[7]。
在猕猴桃中,虽然合成维生素C的主要途径(L-半乳糖途径)的基因没有发生扩张,但是与抗环血酸合成有关的其他基因家族出现了扩张,如Alase(aldonolactonase)、APX(L-ascorbate peroxidase)、MIOX(myo-inositol oxygenase)和维生素C再生途径中的MDHAR(monohydroascorbate reductase)基[JP3]因家族等。可能是由于猕猴桃基因组发生近代的2次WGD事件过程中导致产生了促进维生素C积累的其他基因家族[6]。[JP]
枣果同时具有甜橙和猕猴桃2种积累维生素C的分子机制,即一方面通过L-半乳糖合成AsA的途径得到大幅度加强(类似甜橙),另一方面AsA再生途径中的关键基因家族MDHAR出现极显著扩张(类似猕猴桃)。Liu等分析了AsA的4个合成路径和1个再循环利用途径中的关键酶基因,发现GDP-D-甘露糖3,5差向异构酶和GDP-L-半乳糖磷酸化酶(L-半乳糖途径的2个关键酶)和再循环利用途径中的关键酶(monodehydroascorbate reductase,MDHAR)表现为持续升高的表达水平[9]。
与其他6个蔷薇目基因组水平的比较,在枣果实中 GDP-L-半乳糖磷酸化酶是正向选择基因,而MDHAR表现为显著的扩张。7个蔷薇目树种(包括枣)与1个富含AsA甜橙间的系统发育关系的研究结果表明,有5个主要的MDHAR基因亚家族,亚家族Ⅳ和Ⅴ是枣和蔷薇类特有的。在没有经历近代的WGD事件的物种如草莓、桃、梅、桑椹、枣和甜橙中的基因组中只有1个MDHAR拷贝(亚家族Ⅰ、Ⅱ和Ⅲ),而在经历了近代WGD事件的苹果和梨中却有2个或更多的拷贝[9]。
3.4糖代谢相关的基因
果实的糖代谢对果实风味和色泽的形成以及其他营养成分的代谢具有重要影响,是决定果实品质和商品价值的主要因素。叶片同化的光合产物是果实糖积累的主要来源。苹果、梨等蔷薇科果树,叶片光合产物以山梨醇为主要形态,而非蔷薇科植物以蔗糖为主。
苹果、梨和草莓中,山梨醇代谢途径相关的山梨醇转运酶(SOT)、山梨醇脱氢酶(SDH)和山梨醇-6-磷酸脱氢酶(S6PDH)3个基因家族的基因数目多于非蔷薇类植物。[WTBX][STBX]S6PDH、SDH和SOT[WTBZ][STBZ]基因家族在苹果和梨中发生了扩张,这3个基因家族都属于苹果亚科的特有分支[11]。尽管苹果和梨亲缘关系很近,但[WTBX][STBX]S6PDH[WTBZ][STBZ]基因数目上的差异还是很大的,梨中有4个[WTBX][STBX]S6PDH[WTBZ][STBZ]成员,而苹果有11个。梨中的4个[WTBX][STBX]S6PDH[WTBZ][STBZ]基因分成2簇,分别在5号和2号染色体上,而苹果中只有位于10号染色体上的一个基因簇,其他成员分布于不同的染色体上,表明[WTBX][STBX]S6PDH[WTBZ][STBZ]基因在苹果中的扩张和梨中的收缩可能发生在从共同祖先分化之后。梨中共15个SDH基因,来源相同,集中成簇分布在1号和7号染色体上。在系统发育树上交叉成对,说明SDH基因是经WGD事件进行扩张的。而在苹果中的15个SDH分布较分散,且是不同的基因起源,暗示可能是有潜在的转换事件发生。梨和苹果中都存在树种特异的SOT基因,也说明在从共同的蔷薇类祖先分化后,SOT基因继续扩张。
非蔷薇类的枣果实发育前期,果实中的主要糖类是果糖和葡萄糖,成熟期的枣果中蔗糖和总糖含量在增加,以蔗糖为主。枣中注释到393个与淀粉和蔗糖代谢相关、98个与半乳糖代谢有关、67个与果糖和甘露糖代谢有关、195个与氨基糖和核苷酸糖代谢有关的基因。与其他已测序的蔷薇科果树相比,这些基因家族在枣中都有一定程度上的扩张[9]。
3.5解除休眠的基因
梅、杏、桃、樱桃等李属(Prunus)果树是春季开花较早的果树种类。Zhang等探索了与梅花在低温下打破休眠并开花的分子机制,共鉴定出6个与休眠相关的MADS-box转录因子DAM基因,在基因组中呈串联重复分布。梅中这6个DAM基因是源自一系列复制事件产生的,顺序是[WTBX][STBX]PmDAM1、PmDAM3、PmDAM2、PmDAM5、PmDAM4和PmDAM6[WTBZ][STBZ],DAM基因的这种分子进化模式是李属植物特有的,在桃中也存在,但在苹果和草莓中均没有发现这些串联基因[5],可能与桃、梅、杏、樱桃等李属植物春季开花早于其他大多种类的果树有关。
DAM基因受C-repeat-binding transcription factors(CBF)转录因子的调控。在桃和果梅中[WTBX][STBX]DAM4-DAM6[WTBZ][STBZ]转录起始端上游1 000 bp区域有保守的CBF转录因子位点。在梅中,鉴定出13个CBF同源基因和7个CBF调节子、LEA蛋白。在DAM基因的上游,梅中有比桃更多的[WTBX][STBX]DAM4、DAM5和DAM6[WTBZ][STBZ]的CBF结合位点,并且发现3个新的位点,1个在[WTBX][STBX]DAM1[WTBZ][STBZ]的上游,2个在[WTBX][STBX]DAM6[WTBZ][STBZ]的上游。因此推測DAM基因和过多的CBF结合位点是梅花提早解除休眠的关键因子,使得梅对低温非常敏感,从而导致梅花在早春开花[5]。
3.6自交不亲和基因
自交不亲和性一直是果树分子遗传生物学的研究热点之一,根据花粉不亲和表型的不同遗传方式,植物拒绝自体花粉的再生障碍分为孢子体自交不亲和(sporophytic self-incompatibility,SSI)和配子体自交不亲和(gametophytic self-incompatibility,GSI)[14]。蔷薇科多种果树如梨、苹果、甜樱桃、杏、果梅、李和扁桃等表现出配子体型自交不亲和性,由S位点复等位基因控制,包括2个连锁基因:一个是在雌蕊组织中特异表达的S-RNase基因;一个是花粉中特异表达的SFB(S-haplotype-specific F-box)基因[15-16]。梨基因组研究结果表明,在S-基因座预测到6个SFB候选基因,并且呈现串联重复形式,不同于在苹果和草莓中的随机分布。另外发现梨与苹果在S-基因座上都有高度重复序列,而草莓基因组中没有,重复序列在GSI中的功能还有待于进一步研究[11]。
3.7果实成熟相关的基因
果实成熟过程是果实发育的重要生物学特征,有着重要的经济价值。在番茄中有16个与果实成熟相关的基因,而在柑橘中仅检测到其中的3个:ETR(或NR)、[WTBX][STBX]MADS-RIN和BP0353(或PHYF[WTBZ][STBZ]),可能与呼吸跃变型(番茄)和非呼吸跃变型(柑橘)2种不同的成熟机制有关。尤其是在草莓和番茄果实正常发育成熟过程所必需的MADS-RIN基因,在柑橘果实发育中出现了显著的上调,再次证实了MADS-RIN是2种呼吸类型果实成熟的关键调节子[7]。
3.8抗病基因
抗病性是所有果树植物都关注的重要性状,因此在全基因组解析中也对抗病性相关的基因进行了重点分析。参与植物抗病性的基因主要是R基因,其编码的蛋白具有极高的结构相似性,如亮氨酸拉链(leucine zipper,LZ)、核苷酸结合位点(nucleotide binding site,NBS)、跨膜区域(transmembrane domain,TM)、富含亮氨酸重复(leucine-rich repeats,LRR),以及与果蝇Toll蛋白及哺乳动物白细胞介素-1受体(toll and interleukin-1 receptor,TIR)的细胞外相似区域等。NBS-LRR基因是植物R基因中分布最广和数量最大的1个基因家族。在它们编码蛋白的近N端处存在NBS,而在近C端则存在LRR;而且不同的基因N端还可能包括1个或多个下列2种保守结构:卷曲螺旋(coiled coi1,CC)基序和TIR基序[17]。
在已测序的果树植物研究中主要分析了R基因的9个类型:CC-NBS-LRR、CC-NBS、leucine-rich repeat receptor-like kinase(LRR-RLK)、NBS-LRR、NBS、TIR-CC-NBS-LRR、TIR-CC-NBS、TIR-NBS-LRR和TIR-NBS。其中数量最多的R基因是LRR-RLK、NBS-LRR和TIR-NBS-LRR 3个类型,而TIR-CC-NBS类型占比最少,多数物种中数量是0[5,9]。
由表3看出,在枣与其他11种测序果树基因组NBS-R基因的比较分析中,指出LRR-RLK类型的R基因在每一种果树R基因中的数量均是最高,而与梅中的研究结果不同,占据数量最多的是NBS-LRR或TIR-NBS-LRR;并且对相同果树基因组鉴定出的几种R基因的数量也不同,如苹果基因组在与梅相比时,鉴定出972个R基因[5],在与梨比较时鉴定出1 312(992+320)个[11],而在与枣相比时,共鉴定出 1 511 个R基因[9],可能是与参考的标准序列有关。
尽管不同研究给出的R基因数目有所差异,但物种间横向比较的结果表明,基因组较大的果树植物如苹果基因组中的R基因数目总是相对最高的1个树种,而基因组较小的草莓中R基因的数目是最少的。
另外,已有研究指出梨与苹果中R基因的数目有双重差异。梨中有396个NBS类R基因,占苹果(992个)的399%,并且梨中CC-NBS-LRR基因在数量上超过TIR-NBS-LRRs类型,而苹果则相反。除了NBS基因,梨基因组中有403个LRR-kinase基因和11个其他的CC-LRR-Kinase基因,高于苹果中的320个[11]。
猕猴桃基因组中有261个RLK-LRR,多于葡萄中的232个,与梅中的253个相近[6]。梅中的253个LRR-RLK分为19个基因亚族,其中LRR-Ⅺ和LRR-Ⅻ亚家族呈现显著扩张,类似的扩张也出现在可可和拟南芥基因组。另外,除了R基因,梅花基因组中还存在很多PR基因家族,尤其是[WTBX][STBX]PR10[WTBZ][STBZ]基因家族显著扩张,并且串联分布,[WTBX][STBX]PR10[WTBZ][STBZ]的主要成员串联形成1个小于100 kb的基因簇,而在其他蔷薇科植物中没有发现该基因簇。[WTBX][STBX]PR10[WTBZ][STBZ]基因簇可能与梅花的耐盐、耐旱以及抵抗真菌等抗逆性相关[5]。
在已测序的11个物种中,枣中CC-NBS-LRR类基因是最丰富的。849个R基因中有115个CC-NBS-LRR类基因。17%的枣R基因有1个核苷酸结合配体结构(APAF-1、R-蛋白和CRD-4结构域),是正选择基因,是枣抗病性进化的重要基础[9]。
果树中R基因的进化分析结果表明其有一个共同特点,即R基因在染色体上不是随机分布,而是以串联重复形式排列,并且是集中到某一或几条染色体上。梨中超过30%的R基因是成簇分布的,主要在2、5和11号染色体上[11],有1/3的猕猴桃R基因是集中分布在10个簇内。枣有16%(140个)R基因分布在9号染色体上,表明抗性基因的进化可能与其他植物一样,是串联重复和分化的[6]。
4果树全基因组测序结果的应用与展望
虽然果树植物基因组的测序和组装面临较大的困难,但从已完成全基因组测序的14种果树的分析结果来看,基因组组装的结果质量均较高。通过进一步的生物信息学分析揭示出該物种起源和控制果实性状有关基因的重要基础信息。全基因组测序数据,为世界果树基因组学的研究奠定了重要的信息基础,可为今后其他果树种类的测序研究提供直接的序列数据参考,为同源物种或近缘物种重要性状相关基因的发现、克隆、功能验证和进化分析方面的研究提供极大的便利。
4.1有利于进行转录组测序和关键调控基因挖掘
全基因组序列信息资源的获得,一是可以方便进行同源或近缘果树种类的转录组测序研究,可对不同品种类型、不同组织、不同发育阶段、不同栽培措施处理的材料进行基因表达分析,从而可筛选控制重要性状的关键候选基因。Shi等以发表的桃基因组为参考,对果梅雌蕊败育相关基因进行了转录组分析,初步筛选出与果梅雌蕊发育相关的36个基因[18]。二是通过转录组分析还可进一步提供大量的SSRs和SNP等分子标记,这些序列信息、表达情况以及分子标记将有助于通过遗传图谱进行果树的关键农艺性状QTL定位,并开发与优异性状紧密连锁的分子标记,应于用果树分子标记辅助育种。Wang等参考桃基因组的8个scaffold,成功构建了甜樱桃的高密度遗传图谱,并将与树干直径相关的基因定位到3个连锁群的4个位点[19],为下一步候选基因的筛选、克隆、鉴定奠定了基础。可见,全基因组序列信息资源的获得,将会大大加快同种或近缘果树种类的分子育种进程。
4.2有利于進行比较基因组学研究
多个近缘物种基因组信息的获得,利于在比较基因组学进行更全面、更深入的研究。通过深入比较分析2种植物基因组序列的共线性关系,分析研究植物的起源和进化关系,同时探索控制植物重要性状的重要染色体片段或基因群,也可为重要基因的发现及克隆提供重要参考信息。Illa等通过比较蔷薇科的基因组,构建出蔷薇科家族祖先的假想基因组,并指出小的染色体倒位的发生是蔷薇科进化或蔷薇科内苹果属和李属分化的重要起因[20]。
4.3基因组的3D结构将成为未来研究的热点
线性DNA信息的获得已经是基因组研究的重大进展,但是仅有基因组序列信息还远远不能揭示基因的精准调控和异常表达现象。随着研究的不断深入和全基因组序列图谱的完成,对基因组结构、功能的研究将愈加广阔和深入。随着2009年Hi-C技术的提出和发展,国内外科研人员已开始关注基因组的三维结构。目前在人类基因组与疾病关系的研究方面,已有突出的进展。Tang等利用先进的3D基因组作图策略、3D基因组仿真和超分辨率显微镜,探究了细胞核中的3D基因组结构及其与基因表达和疾病的关系,并第一次鉴别出了染色质蛋白CTCF和cohesin介导的人类基因组高阶和详细的拓扑结构[21]。细胞核内基因组的3D结构影响着基因的表达和DNA复制[22-23]。随着基因组3D结构研究技术的不断开发和发展,果树植物基因组的3D结构研究也将有望迅速开展起来。
随着测序技术、生物信息学分析技术的发展和各种生物学新技术的不断出现,果树基因组学研究将会得到更快更好的发展,果树全基因组及其拓扑结构也将逐渐被一一解析,基因组学的发展必将会解决传统育种方法中存在的盲目性,有望实现通过最有效、最快速的方法获得定点改良和多种优良性状聚合的最优果树品种类型。
[HS2*2][HT8.5H]参考文献:[HT8.SS]
[1][ZK(#]Arumuganathan K,Earle E D. Nuclear DNA content of some important plant species[J]. Plant Molecular Biology Reporter,1991,9(3):208-218.
[2]Jaillon O,Aury J M,Noel B,et al. The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla[J]. Nature,2007,449(7161):463-467.
[3]Velasco R,Zharkikh A,Affourtit J,et al. 2010. The genome of the domesticated apple (Malus×domestica Borkh.)[J]. Nature Genetics,2007,42(10):833-839.
[4]Shulaev V,Sargent D J,Crowhurst R N,et al. The genome of woodland strawberry (Fragaria vesca)[J]. Nature Genetics,2011,43(2):109-116.
[5]Zhang Q X,Chen W B,Sun L D,et al. The genome of Prunus mume[J]. Nature Communication,2012,3(4):1318-1325.
[6]Huang S,Ding J,Deng D,et al. Draft genome of the kiwifruit Actinidia chinensis[J]. Nature Communication,2013,4(4):2640-2649.
[7]Xu Q,Chen L L,Ruan X A,et al. The draft genome of sweet orange (Citrus sinensis)[J]. Nature Genetics,2013,45(1):59-66.
[8]Ming R,VanBuren R,Wai C M,et al. The pineapple genome and the evolution of CAM photosynthesis[J]. Nature,2015,47(2):1435-1441.
[9]Liu M J,Zhao J,Cai Q L,et al. The complex jujube genome provides insights into fruit tree biology[J]. Nature Communication,2014,5:5315-5326.
[10][ZK(#]DHont A,Denoeud F,Aury J M,et al. The banana (Musa cuminata) genome and the evolution of monocotyledonous plants[J]. Nature,2012,488(7410):213-217.
[11]Wu J,Wang Z W,Shi Z B,et al. The genome of pear (Pyrus bretschneideri Rehd.)[J]. Genome Research,2013,23(2):396-408.
[12]Verde I,Abbott A G,Scalabrin S,et al. The high-quality draft genome of peach (Prunus persica) identifies unique patterns of genetic diversity,domestication and genome evolution[J]. Nature Genetics,2013,45(5):487-494.
[13]Osbourn A E,Lanzotti V. Plant-derived natural products[M]. Berlin:Springer Science Business Media,2009:405-431.
[14]McClure B A,Franklin-Tong V. Gametophytic self-incompatibility:understanding the cellular mechanisms involved in “self” pollen tube inhibition[J]. Planta,2006,224(2):233-245.
[15]Zhang S L,Huang S X,Kitashiba H,et al. Identification of S-haplotype-specific F-box gene in Japanese plum (Prunus salicina Lindl.)[J]. Sexual Plant Reproduction,2007,20(1):1-8.
[16]Donia A,Ghada B,Hend B T,et al. Identification,evolutionary patterns and intragenic recombination of the gametophytic self incompatibility pollen gene (SFB) in Tunisian Prunus species (Rosaceae)[J]. Plant Molecular Biology Reporter,2016,34(1):339-352.
[17]Pan Q L,Wendel J,Fluhr R. Divergent evolution of plant NBS-LRR resistance gene homologues in dicot and cereal genomes[J]. Journal of Molecular Evolution,2000,50(3):203-213.
[18]Shi T,Gao Z H,Wang L J,et al. Identification of differentially-expressed genes associated with pistil abortion in Japanese apricot by genome-wide transcriptional analysis[J]. PLoS One,2012,7(10):e47810-47822.
[19]Wang J,Zhang K C,Zhang X M,et al. Construction of commercial sweet cherry linkage maps and QTL analysis for trunk diameter[J]. PLoS One,2015,10(10):e141261-141270.
[20]Illa E,Sargent D J,Girona E L,et al. Comparative analysis of rosaceous genomes and the reconstruction of a putative ancestral genome for the family[J]. BMC Evolutionary Biology,2011,11(1):9-21.
[21]Tang Z,Luo O J,Li X,et al. CTCF-mediated human 3D genome architecture reveals chromatin topology for transcription[J]. Cell,2015,163(7):1611-1627.
[22]Bickmore W A,Van Steensel B. Genome architecture:domain organization of interphase chromosomes[J]. Cell,2013,152(6):1270-1284.
[23]Tjong H,Li W,Kalhor R,et al. Population-based 3D genome structure analysis reveals driving forces in spatial genome organization[J]. Proceedings of the National Academy of Sciences of the United States of America,2016,113(12):1663-1672.
