佐米曲坦缓释及速释固体分散体的制备工艺研究
2017-03-15徐俊闫启东
徐俊+闫启东

[摘要] 目的 制备佐米曲坦缓释和速释固体分散体,提高佐米曲坦生物利用度。 方法 采用固体分散体技术,制备佐米曲坦缓释及速释固体分散体,考察载体种类、用量等对体外释放度的影响。 结果 缓释固体分散体受乙基纤维素(EC)黏度、释放调节剂PEG6000用量、药物与载体用量比例的影响。选择EC(10 cP)和聚乙二醇(PEG)6000混合物为载体材料,其中PEG6000占混合载体总量的40%,控制药物与载体材料比为1∶4,缓释效果较好。采用药物、PEG6000与泊洛沙姆188比例为1∶3.7∶1制备速释固体分散体,药物溶出速率显著提高。 结论 缓释及速释固体分散体能提高药物溶出量,处方合理,具有良好的缓释效果。
[关键词] 佐米曲坦;固体分散体;缓释;速释
[中图分类号] R944.2 [文献标识码] A [文章编号] 1673-7210(2017)01(a)-0016-04
[Abstract] Objective To prepare Zolmitriptan sustained-release and immediate-release solid dispersions (SD) and improve bioavailability. Methods SD technology was used to prepare immediate-release and sustained release SD of Zolmitriptan, the effect of the preparing technology on the in vitro release with the type and quality of carrier material was examined. Results The viscosity of ethyl cellulose (EC) and the dosage of PEG6000 releasing moderator, and the ratio of drug and carrier could impact on the release rates in vitro. When the mixture of EC(10 cP) and PEG6000 as a carrier material to Zolmitriptan was 4∶1, and 40% of the mixed carrier as PEG6000, the sustained-release effect was satisfactory. The dissolution rate were improved significantly to the fast released solid dispersion by using drugs and PEG6000 and poloxamer 188 with a ratio of 1∶3.7∶1. Conclusion The immediate-release and sustained release SD can increase the amount of drug dissolution, the fomulation is reasonable, and the SD using EC and PEG6000 as the carrier has good and stable sustained-release effect.
[Key words] Zolmitriptan; Solid dispersion; Sustained-release; Immediate-release
佐米曲坦(Zolmitriptan)作为二代曲坦类的典型药物,对于偏头痛的治疗具有良好的效果。偏头痛的发作基本为外周和中枢神经血管被激活,引发级联式神经源性炎症[1]。由于神经或血管性功能失调等原因引起偏头痛类疾病,普遍为一侧头部疼痛发作,间或有双侧头部疼痛反复发作等特点[2-3]。曲坦类药物具有良好选择性和强效血清素5-HT1B/1D[4],佐米曲坦通过口服吸收迅速和完全,血浆浓度可以维持4~6 h。其药代动力学呈线性关系的剂量范围为2.5~50 mg,约2.5 h的消除半衰期,生物利用度大约为40%,佐米曲坦和它的代谢物与血浆蛋白结合率较低[5],约为25%。在售药品药物近40%属于难溶性药品,而在研药物中难溶性药物比例更高[6]。由于溶解性的限制,一些具有药理活性的新化合物没有办法被制成相应制剂并在临床应用。学者们对药物的缓释制剂制备进行探索研究,做出了卓有成效的工作[7-12]。荣毅等[13-14]对手性药物选择性释药进行了较全面的研究。徐俊等[15]报道了琥珀酸舒马曲坦固体分散体颗粒的研究探索,取得了良好的效果。张伟等[16-17]报道了口服固体制剂溶出度的测定方法。
本研究采用固体分散体(solid dispersion,SD)技术制备佐米曲坦缓释(slow-release,SR)及速释(quick-release,QR)SD,针对体外释放度考察了载体种类、用量等因素的影响,进行了工艺条件的探索优化,旨在制备释放性能较优的SRSD和QRSD。SRSD具有延缓释放和提高藥生物利用度的效果;QRSD具有提高药物释放速度和生物利用度的作用。作为固体制剂,佐米曲坦SRSD和QRSD可以单独使用,也可以组合使用,对于提高药物的生物利用度有所提高,亦可调节药物的释放速度。若将SR和QR两种SD按照一定的比例组合,药物溶出时间缩短,快速起效,不仅能延缓释放时间提高药物应用的效果,而且改善了现有制剂起效时间相对较长等缺点。
1 仪器与试药
紫外分光光度计:UV-1801型,北分瑞利仪器有限公司;溶出度测定仪:RC-8D型,天津光学仪器有限公司;旋转蒸发仪:RE-52AA型,上海亚荣生物仪器有限公司。
佐米曲坦(批号:130401,湖北威德利化学科技有限公司);佐米曲坦标准品(USP-1727009,美国药典USP标准品);乙基纤维素(型号:EC 10~100 cP,美国陶氏集团);泊洛沙姆188(德国BASF公司);聚乙二醇(型号:PEG6000,上海强顺化学品有限公司);十二烷基硫酸鈉(SDS)(上海强顺化学品有限公司),以上试剂皆为分析纯。
2 方法与结果
2.1 SD与物理混合物制备
采用溶剂法和溶剂熔融法[18]制备佐米曲坦QRSD和SRSD。SRSD以EC及PEG为载体,以延缓所制备的剂型药物的释放度;QRSD以PEG6000及泊洛沙姆188为载体进行制备。
2.1.1 佐米曲坦SRSD制备 精密称取一定量的佐米曲坦,加入4倍量(W/V,g/mL)的混合有机溶剂(丙酮∶无水乙醇=2∶1)溶解,待溶液澄清;取一定量的混合载体,精密称定,加入无水乙醇溶剂,体积控制在4~7倍(W/V,g/mL)的药物量。在低温(<50℃)加热条件下搅拌溶解;佐米曲坦溶液加入载体溶液中混合,于65℃搅拌充分混匀,旋转蒸发仪中将溶剂挥干后冷却至室温,固体粉碎至粗颗粒,于真空干燥器(温度30℃,压强5 kPa以下)干燥24 h,粉碎过40~80目筛,粉末干燥避光保存。
2.1.2 佐米曲坦QRSD制备 精密称取一定量的佐米曲坦,加入4倍量(W/V,g/mL)的混合有机溶剂(丙酮∶无水乙醇=2∶1)溶解,待溶液澄清;取一定量的混合载体,精密称定,加入无水乙醇溶剂,体积为4倍(W/V,g/mL)的药物量。在低温(<50℃)加热条件下搅拌溶解;佐米曲坦溶液加入载体溶液中混合,于60℃搅拌充分混匀,旋转蒸发仪中将溶剂挥干后冷却至室温,固体粉碎至粗颗粒,于真空干燥器(温度30℃,压强5 kPa以下)干燥24 h,粉碎过40目筛,粉末干燥避光保存。
2.1.3 佐米曲坦物理混合物制备 取上述SD药物与所选载体处方量,直接研磨混匀,过40目筛即得产品。
2.2 溶出度试验
取制备的佐米曲坦SD粉末10 mg,溶出度测定方法参照中国药典[19]操作。以0.5%的SDS溶液900 mL为释放介质,在控制温度为(37.0±1.0)℃、转速为50 r/min条件下,分别于1、2、5、10、15、20、30、45、60 min对QRSD取样6.0 mL;于0.5、1、2、4、6、8、10、12 h对SRSD取样6.0 mL,同时补充6.0 mL的溶出介质,用0.8 μm微孔滤膜过滤,以含0.5%的SDS溶液的空白溶剂对照,滤液于225 nm波长处测定吸光度,计算不同时间药物的累积释放度(Q,%),绘制溶出曲线。
2.3 SR佐米曲坦SD的筛选与优化
用单一指标考察SR效果不显著,根据SD常用的制备配方比例和优化工艺探索方面考虑,采用单因素法进行筛选和优化。
2.3.1 载体中EC种类考察 选择四种黏度规格的EC(10、20、45、100 cP),按照佐米曲坦与混合载体量总量比例为1∶4,释放调节剂PEG6000与EC比例为1∶2,制得SD,考察EC黏度对药物缓释的影响,结果见图1。从结果可见,四种EC所制得SD均有缓释作用,但EC的黏度越大,水化速度越快,越容易形成凝胶层控制药物释放,导致药物释放越慢,且黏度较高的EC(45、100 cP)12 h内无法完全释放药物。黏度为10 cP或20 cP的EC均有较好的释放效果,其中EC(10 cP)较EC(20 cP)释放作用稍好。
2.3.2 释放调节剂比例考察 选用EC(10 cP)为SRSD载体,以PEG6000为释放调节剂,选用不同用量,使PEG6000分别占混合载体总量的10%、20%、30%、40%、50%,按照佐米曲坦与混合载体总量比例为1∶4,制得SD,考察PEG6000对药物缓释的影响,结果见图2。从结果可见,随着PEG6000用量增大,释放速率逐渐提高,且PEG6000在载体中含量低于30%不能完全释放。PEG6000占混合载体总量的30%~40%,缓释作用较好,12 h内可持续释放,其中PEG6000占混合载体总量40%时释放作用更好。
2.3.3 药物与载体比例考察 选用EC(10 cP)为SRSD载体,以PEG6000为释放调节剂,释放调节剂PEG6000占混合载体总量的40%,调整药物与载体量比例分别为1∶2、1∶3、1∶4、1∶5、1∶6、1∶7,制得SD,考察药物与载体量比例对药物缓释的影响,结果见图3。从结果可见,随着载体用量增加,释放速率逐渐提高,但载体用量需选择适中,不是越多越好,当载体用量较大时,因释放阻力增大,导致12 h内不能完全释放。药物与载体用量比例为1∶3、1∶4、1∶5时,12 h内缓释作用较好。
2.4 QRSD的筛选与优化
2.4.1 载体的选择和优化 采用单一载体按照佐米曲坦与载体总量比例为1∶4,制备SD,其溶出曲线与同比例的PEG6000与佐米曲坦的物理机械混合物、泊洛沙姆188与佐米曲坦的物理机械混合物进行比较,结果见图4。从图4可见,PEG6000载体和泊洛沙姆188载体制备的SD,其溶出效果都有相对明显的改进,并且效果相近。
2.4.2 混合载体选择和优化 PEG6000与泊洛沙姆188作为单一载体制备SD,其溶出效果比较接近,故优先使用PEG6000可降低成本。相对于PEG6000载体,泊洛沙姆188载体具有强的增溶作用,为了提高溶出的效果和实践,进行适量的添加。
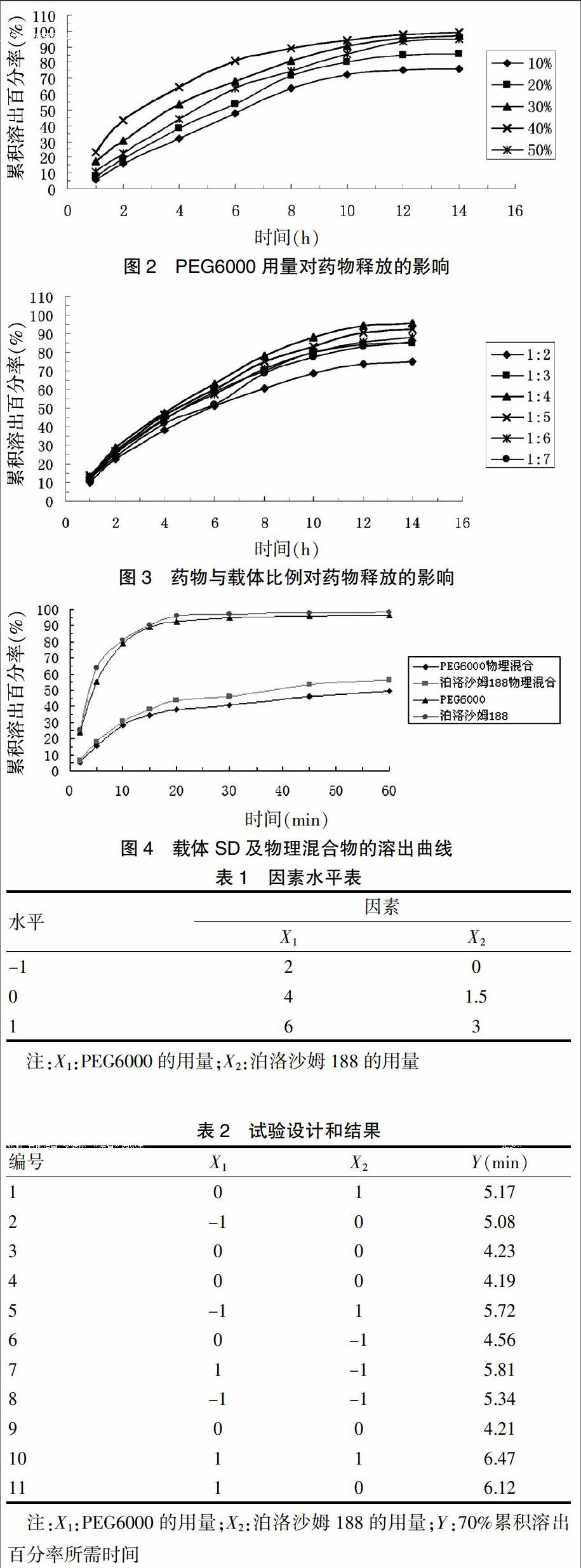
优化结果显示,载体和药物比例控制在2∶1~6∶1之间SD溶出较好。在单因素预实验基础上,以PEG6000(X1)、泊洛沙姆188(X2)为自变量,以70%累积溶出百分率所需的时间(Y)作为响应值,采用Box-Behnken响应面法[20]分析进行二因素实验设计,见表1。考察PEG6000中添加泊洛沙姆188制备SD的溶出效果,见表2。以佐米曲坦原料用量为1;因素X1范围:2~6;因素X2范围:0~3。求得回归方程为:Y=8.51 982-2.184 56×X1-0.488 60×X2+0.023 333×X1×X2+0.292 24×X12+0.192 87×X22。拟合方程的相关系数R2=0.9728,X1、X2对时间的影响均比较显著。
通过模型方程作出响应曲面图,见图5。从图5可知响应曲面较陡,说明PEG6000用量与泊洛沙姆188用量的交互作用影响较大,宜采用PEG6000和泊洛沙姆188混合载体制备QRSD。
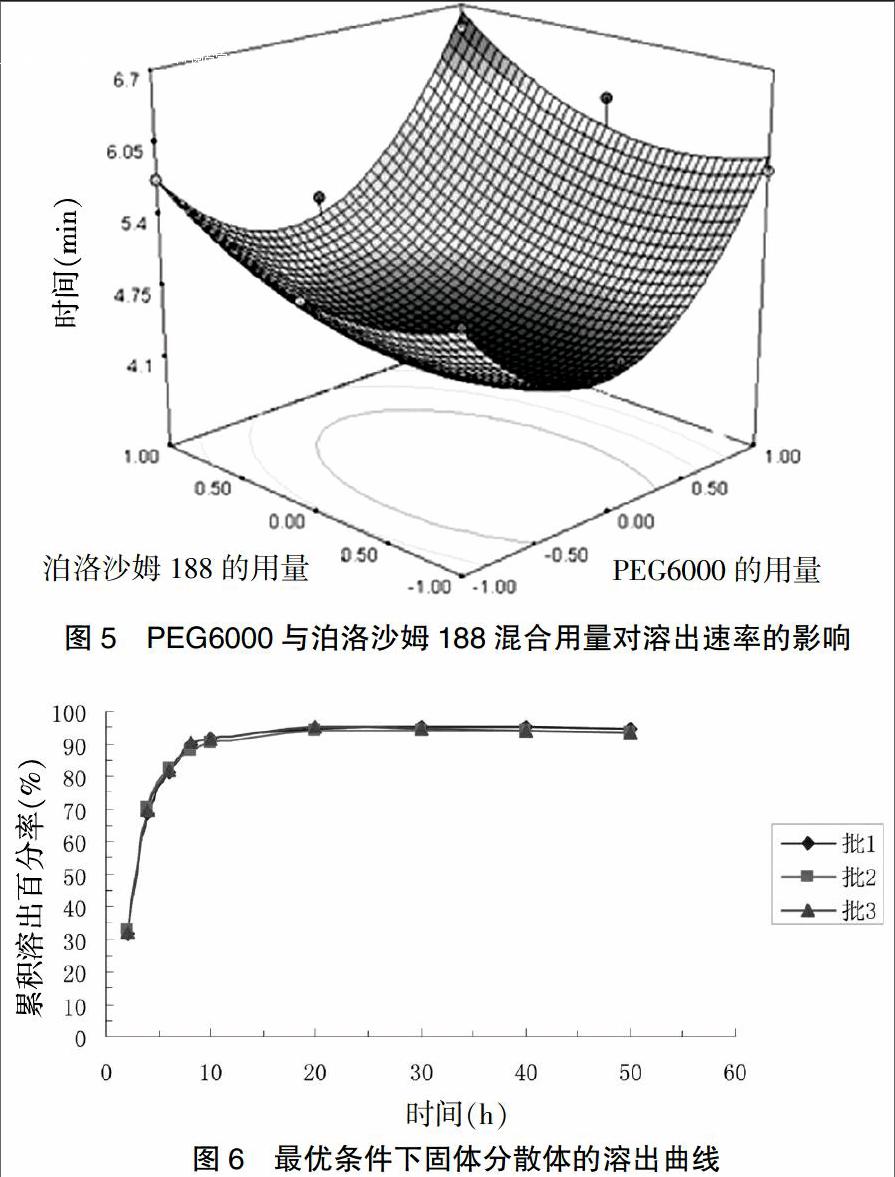
根据软件分析,结果预测值X1=3.7,X2=1.04时,Y=4.22791。按預测值制备三批佐米曲坦QRSD,进行溶出速率检测,结果70%累积溶出达到时所需时间约为4.5 min,与预测值比较接近。见图6。
3 讨论
缓释载体的用量(含释放调节剂的用量)与药物的比例影响SRSD的溶出速度及溶出总量。采用EC和PEG6000为混合载体制备SRSD的释放速率延缓,药物与混合载体比例控制在1∶3~1∶5时缓释作用较好。混合载体中PEG6000比例太低导致释放不完全,PEG6000比例太高释放过快,PEG6000占混合载体总量的30%~40%,12 h内缓释作用较好。
王美荣等[21]报道,使用CH3OH和CH2Cl2为混合溶剂,以PVP K30为药物载体来制备SD。但因溶剂的毒性大、载体吸湿性过高,不利于SD的生产操作。采用PEG6000和泊洛沙姆188混合载体制备QRSD,释放效果较好,相对较快且基本释放完全,且PEG6000价格较低,节约成本。混合载体中PEG6000和泊洛沙姆比例为3.7∶1,溶出速率有显著提高。不足之处在于其中混合载体中EC的黏度会影响释放效果,黏度过高形成凝胶层释放过慢。也可能会受体内释药环境pH值和其他因素的影响,缓控释效果有所不同。
[参考文献]
[1] 何增柳,董兰真,张艺馨,等.偏头痛与代谢综合征的相关性研究[J].重庆医科大学学报,2015,40(5):699-702.
[2] 陈明波,刘志明,朱红雯.偏头痛发病机理及临床治疗进展[J].中外医疗,2011,30(21):192.
[3] 张勇,付彩红,任毅,等.偏头痛的功能MRI研究进展[J].磁共振成像,2014,5(5):396-400.
[4] 郑丽玲,高旭光.偏头痛发作期治疗的研究进展[J].中国全科医学,2013,16(9):3167-3169.
[5] Dixon R,Warrander A. The clinical pharmacokinetics of Zolmitrptan [J]. Cephalagia,1997,17(8):15-20.
[6] 顾艳,何军,卞玮,等.注射纳米混悬剂制备及表征的研究进展[J].中国医药工业杂志,2016,47(4):471-476.
[7] 李菲,张洁,王建筑,等.尼莫地平缓释滴丸的制备及其体外释药机制的探讨[J].中国新药杂志,2016,25(20):2360-2364.
[8] 袁春平,梁华娟,林碧珊,等.伪麻非索缓释胶囊与原研缓释片体外释放行为一致性评价[J].中国医药工业杂志,2016,47(6):2755-2760.
[9] 王磊,丁四海,刘秀茹,等.对乙酰氨基酚复合微囊缓释栓的制备及体外释放度研究[J].中国医院药学杂志,2016,36(9):714-718.
[10] 高捷,朱淼,沃联群,等.盐酸拉贝洛尔时控缓释微丸的研制[J].中国医药导报,2016,13(19):26-30.
[11] 胡献跃,郑一美.盐酸帕罗西汀缓释片的研制[J].中国药业,2016,25(17):31-34.
[12] 黄韵然,陈玲,卢克伟,等.盐酸伪麻黄碱/盐酸非索非那定复方缓释胶囊的研究.Ⅲ.体内外评价[J].中国医药工业杂志,2016,47(5):582-586.
[13] 荣毅,张亮,俞文英,等.基于分子印迹技术的手性药物拆分及对映体选择性释药系统研究进展[J].中国医药工业杂志,2014,45(4):381-386.
[14] Yin J,Cui Y,Yang G,et al. Molecularly imprinted nanotubesfor enantioselective drug delivery and controlled release [J].Chem Commun (Camb),2010,46(41):7688-7690.
[15] 徐俊,闫启东,石雷.琥珀酸舒马曲坦固体分散体颗粒溶出度的测定[J].中国医院药学杂志,2016,45(5):972-974.
[16] 张伟,刘建芳,赵彩霞.口服固体制剂溶出度测定方法的研究与应用进展[J].中国医药导报,2016,13(3):48-50.
[17] Klein S,Garbacz G,Pislar M,et al. The role of individual gastric emptying of pellets in the prediction of diclofenacin vivo dissolution [J]. J Control Release,2013,166(3):286-293.
[18] 陆彬.药物新剂型与新技术[M].北京:人民卫生出版社,2003:12-15.
[19] 国家药典委员会.中国药典[M].二部.北京:中国医药科技出版社,2010:附录84.
[20] 孟戎茜,王曼,郭聪.布洛芬滴丸的成型工艺及体外溶出度研究[J].中国药房,2016,27(25):3558-3561.
[21] 王美荣,王利珍,林建英.坎地沙坦固体分散体的制备及溶出度、稳定性研究[J].中国现代应用药学,2011,28(7):654-658.
(收稿日期:2016-09-30 本文编辑:王红双)
