含氧溶液中毒砂氧化溶解的XAFS研究
2016-12-23张国庆张明美王少锋贾永锋
张国庆 张明美 王少锋 贾永锋
1(中国科学院沈阳应用生态研究所 污染生态与环境工程重点实验室 沈阳 110016)2(中国科学院大学 北京 100049)
含氧溶液中毒砂氧化溶解的XAFS研究
张国庆1,2张明美1王少锋1贾永锋1
1(中国科学院沈阳应用生态研究所 污染生态与环境工程重点实验室 沈阳 110016)2(中国科学院大学 北京 100049)
毒砂的氧化溶解是环境As污染的重要来源之一。本研究对毒砂的氧化反应过程及产物进行了研究。考察了不同参数:pH、溶解氧(Dissolved Oxygen, DO)对毒砂氧化反应的影响,利用X射线吸收精细结构(X-ray Absorption Fine Structure, XAFS)分析了毒砂氧化过程中As的形态转化。XAFS光谱学数据表明,pH=4时,毒砂发生较强的氧化反应,释放的As(V)被氧化过程中形成的铁氧化物吸附;pH=7时,毒砂氧化释放的As(III)被铁氧化物吸附,表面存在少量的类似雄黄(AsS)的物质;而pH=9时,毒砂几乎不发生氧化,表面只存在少量的硫代亚砷。结果表明,pH是影响毒砂氧化的重要因素。中碱性条件下,毒砂氧化反应释放出的Fe快速形成了Fe的氧化物沉淀,包裹在毒砂的表面,阻碍毒砂继续氧化。
毒砂,氧化反应,铁氧化物,X射线吸收精细结构
水土砷污染对环境和人类健康造成了严重的危害,是世界性的环境问题[1-3]。自然环境中的As主要来源于人为砷排放与自然砷释放,水体中砷的自然释放过程主要是Fe、Mn氧化物上的As的解吸和含砷矿物的氧化溶解[4-5]。自然界中的含As矿物主要有毒砂、雄黄、雌黄以及含As黄铁矿等。毒砂和含砷黄铁矿的氧化溶解,产生大量的高浓度含As酸性废水[6-8]。导致大量的As排放到水体中[9-13]。对生态环境造成了严重的危害,如西班牙中部和中国卡林型金矿,由于毒砂和含砷黄铁矿的氧化溶解导致非常严重的地下水As污染[14]。
温度、pH、溶解氧(Dissolved Oxygen, DO)、Fe(III)浓度、微生物等是影响毒砂氧化溶解的主要因素[15-18],前人主要研究了pH、颗粒大小的影响[19-20],以及液相化学或是利用X射线光电子能谱分析(X-ray Photoelectron Spectroscopy, XPS)来进行毒砂的表面光谱学研究[20-22]。而对毒砂氧化过程及As、Fe的最终形态及分配研究很少。
前期毒砂氧化溶解研究认为,在不同的实验参数和有限的实验条件下,毒砂的氧化动力学研究中,一些基础过程的结论仍存在分歧[23-24]。例如:毒砂表面S的形成、化学计量溶解、氧化速率的变化、氧化速率与DO之间的相关性以及出现不规则的速率与Arrhenius的行为。
本文主要研究了在不同的氧化条件(pH、DO)下,毒砂的氧化动力学。分析了中碱性条件下,Fe氧化物沉淀对毒砂氧化溶解的影响,并采用X射线吸收精细结构(X-ray Absorption Fine Structure, XAFS)分析了毒砂氧化溶解过程中As的形态转化。研究的结果可为含砷矿物的氧化溶解导致的As释放而引起的环境危害提供一定的理论依据。
1 材料与方法
1.1毒砂的制备
将块状毒砂经过研磨后过100目筛,乙醇中洗涤10 min,之后用超声清洗三次,每次1 min,去除晶体表面的细小颗粒,最后用去离子水清洗三次,丙酮清洗两次,晾干后储存于干燥器中待用。
1.2毒砂的氧化实验
毒砂氧化实验使用生物反应器Fermac320(英国Electrolab),可自动记录并精确控制pH、DO、温度。实验时,称量0.8 g毒砂粉末放入反应器中,加入3.5 L的蒸馏水,以3.3 g·L-1的KCl作为电解质,调节不同的反应参数(pH、DO)进行实验。分别进行了pH=4、7、9实验,每组pH实验进行4 d,每天的取样时间为0 h、2 h、4 h、6 h、8 h、10 h、12 h、14 h、24 h。通入空气和氮气调DO,每24 h调节一次,该仪器通过气体百分比控制体系中的DO,调至90% DO、70% DO、50% DO、30% DO,DO浓度分别为7.3 mg·L-1、5.8 mg·L-1、4.2 mg·L-1、2.5 mg·L-1。体系温度保持在25 ºC,转速为400 r·min-1。pH计与DO电极采用标准方法定期校正。
每个取样点取样两次,每次取5 mL样品经过0.22 μm滤膜过滤,测定液相中As、Fe含量。另取1 mL样品,加入1 mL HCl (1 mol·L-1)溶解无定型铁氧化物,0.5 h后用0.22 μm滤膜过滤后,测定溶液中的As、Fe含量。
1.3砷、铁的分析方法
砷含量测定:采用原子荧光光度计分析液相中As(T)和As(III)的含量,仪器检测线为0.01 μg L-1,分析精度为±5%。
As(T)浓度测定时,预先向样品中加入1 mL还原剂(5%硫脲和5%抗坏血酸的混合液),用5% HCl定容至5 mL,放置至少6 h后,泵入原子荧光光谱仪测定,以5% HCl作为载液,硼氢化钾溶液(0.03% NaOH和2%硼氢化钾的混合液)作为还原剂[25]。
As(III)浓度测定时,直接用柠檬酸-柠檬酸三纳缓冲液(pH约为5)定容,泵入原子荧光光谱仪测定,柠檬酸-柠檬酸三纳缓冲液作为载液,硼氢化钾溶液作为还原剂[26]。
铁含量测定:将待测样品用1% HNO3溶液稀释后,采用原子吸收分光光度计(AA240,美国Varian)测定,检出限为0.1 mg·L-1。
1.4 X射线衍射分析
X射线衍射分析(X-ray Diffraction Analysis, XRD)谱图在D/MAX 2400型X射线衍射仪上测定。实验采用Cu Kα射线(λ=0.5418 nm),石墨单色器,工作电压56 kV,工作电流182 mA,发散狭缝1º,接受狭缝1º,室温下收集10º-90º (2θ)衍射数据。
1.5同步辐射表征
As的K边XAFS光谱在北京同步辐射装置XAFS站获得。测样时,将毒砂氧化前后的固体粉末样品均匀地凃在Kapton胶带上,固定在样品架上,室温下采集,As的K吸收边能量为11867 eV, XAFS光谱能量采集范围11667-12667 eV,X射线吸收近边结构(X-ray Absorption Near Edge Structure, XANES)光谱段的步长为0.5 eV,高浓度样品采用透射模式测定,低浓度样品采用荧光模式测定,每个样品重复扫描三次。As标准物为砷酸钠(Na3AsO4·12H2O)、亚砷酸钠(Na3AsO3)、硫代亚砷酸钠(Na3AsO2S)、硫代砷酸钠(Na3AsO3S)、雌黄(As2S3)、雄黄(AsS)和毒砂(FeAsS)。XAFS光谱数据的线性拟合(Linear Combination of Fits, LCF)采用(D)Athena(5.12.3)进行分析。
2 结果与分析
2.1固体表征
XRD结果如图1所示。实验所采用的样品的主要矿物形式为FeAsS (PDF#85-1723)。样品比表面积和平均孔径分别为1.64 m2·g-1和1.63 nm。

图1 毒砂的XRD表征Fig.1 XRD pattern of FeAsS.
2.2酸性条件下毒砂的氧化溶解
如图2所示,pH=4时,毒砂表面的细小颗粒首先被氧化[27],导致体系盐酸可提取态的As、Fe的起始值约为2 mg·L-1和5 mg·L-1。在90% DO时,As和Fe的释放量都随时间增加;在70% DO阶段,As(T)和Fe(T)释放量呈增加趋势,而As(III)在固相和液相中已达到平衡;50% DO和30% DO阶段,毒砂氧化过程中固液相As(III)含量逐渐降低,而As(T)和Fe(T)含量逐渐增加。图2中As(III)a、As(T)a、Fe(T)a分别代表液相中As(III)含量、总砷含量、总铁含量(mg·L-1),As(III)s、As(T)s、Fe(T)s代表固相中盐酸可提取As(III)含量、总砷含量、总铁含量(mg·kg-1)。
如图2(a)所示,由于在每个DO转换点上,毒砂氧化释放的As含量短时间内先下降后上升,而液相中As没有出现类似现象,说明固相中盐酸可提取态As含量降低导致As(T)暂时降低。这可能是由于DO交换点,Eh值突然下降,使得毒砂释放S2-,与As反应形成AsS或As2S3沉淀,而AsS、As2S3不溶于HCl,因此造成盐酸可提取As含量降低,但随着Eh逐渐增加,AsS、As2S3被氧化,盐酸可提取态砷含量迅速增加,发生的反应如下:

之后As(III)再被O2氧化:
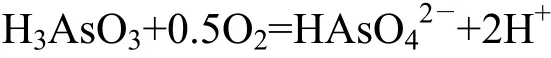
由图2(b)中固相和液相Fe(T)的差值可知,在pH=4时,毒砂氧化过程中形成了少量的Fe的氧化物沉淀,但并没对As的释放造成影响。

图2 pH=4毒砂氧化过程中液相与盐酸可提取As (a)和Fe (b)的含量变化Fig.2 Variation of the aqueous and HCl extractable As (a) and Fe (b) concentration at pH=4.
2.3中性条件下毒砂的氧化溶解
pH=7时,DO的调节时间与pH=4时相同。实验期间,DO波动的相对偏差小于5%,pH的范围在6.7-7.2,实验过程中氮气和空气的交替通入会导致Eh突然降低后升高,Eh在-75-25 mV内变化。
如图3所示,在90% DO阶段,在反应初8 h内,盐酸可提取态的As(III)和As(T)含量快速增加,从约0.2 mg·L-1和1 mg·L-1迅速增加到1.1 mg·L-1和3.8 mg·L-1。随后盐酸可提取态的As(III)呈缓慢升高趋势,而盐酸可提取的As(T)、液相As(T)和As(III)则几乎达到平衡。

图3 pH=7毒砂氧化过程中液相与盐酸可提取As (a)和Fe (b)的含量变化Fig.3 Variation of the aqueous and HCl extractable As (a) and Fe (b) concentration at pH=7.
与酸性条件下(pH=4)的变化类似,在DO切换点,中性条件下盐酸可提取的As(T)含量也有所降低,但降低幅度小于酸性条件,表明在毒砂表面可能形成了As的硫化物沉淀,但AsS和As2S3在酸性条件下较为稳定,而在中碱性条件下不稳定,因此产生量会低于酸性条件。
从毒砂的氧化比例看,中性条件下毒砂的氧化量低于酸性条件,这也是盐酸可提取态As(T)降低幅度较小的原因。中性条件下,毒砂反应释放的Fe(II)很快被氧化为Fe(III)并形成Fe氧化物沉淀,所以测得液相中Fe(T)的含量低于仪器的检测限,固相中Fe(T)也在24 h内迅速升高,之后在4-6.8 mg·L-1内波动。
2.4碱性条件下毒砂的氧化溶解
pH=9实验中,DO切换时间与pH为4、7相同,且可较为精确地控制,pH的变化范围在8.9-9.1,Eh在-30-46 mV内变化。碱性条件下(pH=9),毒砂氧化释放的Fe完全氧化生成Fe的氧化物沉淀,此时液相中Fe(T)的含量低于仪器检测限。
如图4所示,毒砂在碱性条件下生成的Fe氧化物沉淀最大值为5 mg·L-1,比pH=7小,反应过程中Fe的变化不大。在90% DO阶段,固相和液相中的As(III)和As(T)在氧化初期的5 h内随时间迅速增加并达到一个平台。当DO降至70%时,盐酸可提取态As(III)和As(T)以及液相As(III)含量开始降低。在50% DO-30% DO中,液相As(III)和As(T)都逐渐达到平衡,固相中的As(III)出现大幅度降低,As(T)开始升高,大量的As(III)被氧化生成As(V)。

图4 pH=9毒砂氧化过程中液相与盐酸可提取As (a)和Fe (b)的含量变化Fig.4 Variation of the aqueous and HCl extractable As (a) and Fe (b) concentration at pH=9.
2.5毒砂氧化过程中As的形态转化
不同pH条件毒砂氧化释放的As形态以及固相和液相之间的分配不同。pH=4时,没有形成过多的Fe的氧化物沉淀,对As的释放几乎没有影响,As和Fe按照化学计量释放。
与前人研究[16,19,30]得到的酸性条件下毒砂的氧化机制相同,按照下列反应方程式:

然后H3AsO3氧化成HAsO42-和H2AsO4-:
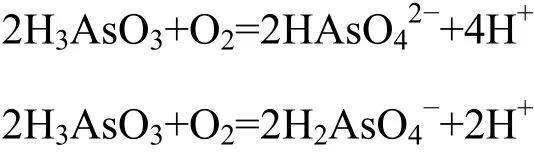
pH=4时,固相和液相中As(T)、Fe(T)含量相差很少,说明铁氧化物吸附的As较少;pH=7时,毒砂氧化快速达到了平衡,释放的Fe完全转化生成铁氧化物。液相中As(III)和As(T)含量几乎相同,说明毒砂氧化释放出的As均为As(III)。固相中的As(III)比液相多,所以部分的As(III)被铁氧化物吸附,同时吸附的As(III)被氧化为As(V)。pH=9时,固相和液相As的差值表明铁氧化物沉淀吸附了大量的As,48 h后固相中出现As(III)和As(T)的较大差值,表明了吸附在铁氧化物上的As(III)被氧化为As(V)。
如图5和表1所示,pH=9时,毒砂几乎不发生氧化,表面只存在少量的硫代亚砷。pH=7时,毒砂氧化强度不大,毒砂氧化释放的As(III)被氧化过程形成的铁氧化物吸附,表面存在少量的类似雄黄(AsS)的物质。而pH=4,从图5可以明显看出,有As(V)生成,发生较强的氧化反应,同时生成的铁氧化物吸附了As(V),与pH=7相比,表面形成了较多类似雄黄的物质,这就与前面pH=4实验中,在DO 转换点上出现As突然下降的现象吻合,是形成了类似雄黄的物质导致的。

图5 氧化毒砂反应产物的同步辐射表征与线性拟合Fig.5 Linear combination fitting of XANES spectra of oxidized arsenopyrite.

表1 毒砂氧化反应产物的As K边XANES的LCF拟合结果Table 1 Products of arsenopyrite oxidization deduced from LCF results of As K-edge spectra.
3 结语
毒砂的氧化溶解是环境As污染的重要来源之一,而pH是影响毒砂氧化的重要因素。酸性条件下,毒砂易发生较强烈的氧化反应,导致大量As释放到液相中;中碱性条件下,毒砂反应释放出的Fe快速形成了Fe的氧化物沉淀,包裹在毒砂的表面,阻碍毒砂继续氧化。酸性条件下毒砂的氧化,产物倾向于进入液相中,因此采矿过程中产生的高浓度的酸性矿山废水,可导致大量砷进入土壤、地表或地下水体,从而对生态环境造成严重危害。
1 Aposhian H V, Zakharyan R A, Avram M D, et al. A review of the enzymology of arsenic metabolism and a new potential role of hydrogen peroxide in the detoxication of the trivalent arsenic species[J]. Toxicology & Applied Pharmacology, 2004,198(3): 327-335
2 Bunnell J E, Finkelman R B, Centeno J A, et al. Medical geology: a globally emerging discipline[J]. Geologica Acta, 2007,5(3): 273-281
3 Rossman T G, Uddin A N, Burns F J. Evidence that arsenite acts as a cocarcinogen in skin cancer[J]. Toxicology and Applied Pharmacology, 2004,198(3): 394-404
4 Casiot C, Leblanc M, Bruneel O, et al. Geochemical processes controlling the formation of As-rich waters within a tailings impoundment (Carnoules, France)[J]. Aquatic Geochemistry, 2003,9(4): 273-290
5 Pfeifer H R, Haussermann A, Lavanchy J C, et al. Distribution and behavior of arsenic in soils and waters in the vicinity of the former gold-arsenic mine of Salanfe, Western Switzerland[J]. Journal of Geochemical Exploration, 2007,93(3): 121-134
6 Acero P, Ayora C, Torrento C, et al. The behavior of trace elements during schwertmannite precipitation and subsequent transformation into goethite and jarosite[J]. Geochimica Et Cosmochimica Acta, 2006,70(16): 4130-4139
7 Asta M P, Cama J, Ayora C, et al. Arsenopyrite dissolution rates in O-2-bearing solutions[J]. Chemical Geology, 2010,273(3-4): 272-285
8 Sanchez-Rodas D, Gomez-Ariza J L, Giraldez I, et al. Arsenic speciation in river and estuarine waters from southwest Spain[J]. Science of the Total Environment, 2005,345(1-3): 207-217
9 Frau F, Ardau C. Geochemical controls on arsenic distribution in the Baccu Locci stream catchment (Sardinia, Italy) affected by past mining[J]. Applied Geochemistry, 2003,18(9): 1373-1386
10 Lazareva E V, Shuvaeva O V, Tsimbalist V G. Arsenic speciation in the tailings impoundment of a gold recovery plant in Siberia[J]. Geochemistry Exploration Environment Analysis, 2002,2(3): 263-268
11 Lee P, Kang M J, Choi S H, et al. Sulfide oxidation and the natural attenuation of arsenic and trace metals in thewaste rocks of the abandoned Seobo tungsten mine, Korea[J]. Applied Geochemistry, 2005,20(9): 1687-1703
12 Smedley P L, Kinniburgh D G. A review of the source, behaviour and distribution of arsenic in natural waters[J]. Applied Geochemistry, 2002,17(5): 517-568
13 Williams M. Arsenic in mine waters: an international study[J]. Environmental Geology, 2001,40(3): 267-278
14 Hernaandez-Garcia M E, Custodio E. Natural baseline quality of madrid tertiary detrital aquifer groundwater (Spain): a basis for aquifer management[J]. Environmental Geology, 2004,46(2): 173-188
15 Breed A W, Harrison S T L, Hansford G S. A preliminary investigation of the ferric leaching of a pyrite/arsenopyrite flotation concentrate[J]. Minerals Engineering, 1997,10(9): 1023-1030
16 Walker F P, Schreiber M E, Rimstidt J D. Kinetics of arsenopyrite oxidative dissolution by oxygen[J]. Geochimica Et Cosmochimica Acta, 2006,70(7): 1668-1676
17 Craw D, Falconer D, Youngson J H. Environmental arsenopyrite stability and dissolution: theory, experiment, and field observations[J]. Chemical Geology, 2003,199(1-2): 71-82
18 McArthur J M, Ravenscroft P, Safiulla S, et al. Arsenic in groundwater: testing pollution mechanisms for sedimentary aquifers in Bangladesh[J]. Water Resources Research, 2001,37(1): 109-117
19 McKibben M A, Tallant B A, del Angel J K. Kinetics of inorganic arsenopyrite oxidation in acidic aqueous solutions[J]. Applied Geochemistry, 2008,23(2): 121-135
20 Nesbitt H W, Muir I J. Oxidation states and speciation of secondary products on pyrite and arsenopyrite reacted with mine waste waters and air[J]. Mineralogy and Petrology, 1998,62(1-2): 123-144
21 Mikhlin Y L, Romanchenko A S, Asanov I P. Oxidation of arsenopyrite and deposition of gold on the oxidized surfaces: a scanning probe microscopy, tunneling spectroscopy and XPS study[J]. Geochimica Et Cosmochimica Acta, 2006,70(19): 4874-4888
22 Richardson S, Vaughan D J. Arsenopyrite - a spectroscopic investigation of altered surfaces[J]. Mineralogical Magazine, 1989,53(370): 223-229
23 Corkhill C L, Vaughan D J. Arsenopyrite oxidation - a review[J]. Applied Geochemistry, 2009,24(12): 2342-2361
24 Lengke M F, Sanpawanitchakit C, Tempel R N. The oxidation and dissolution of arsenic-bearing sulfides[J]. Canadian Mineralogist, 2009,47(3): 593-613
25 Xu L, Zhao Z, Wang S, et al. Transformation of arsenic in offshore sediment under the impact of anaerobic microbial activities[J]. Water Research, 2011,45(20): 6781-6788
26 Wang S, Xu L, Zhao Z, et al. Arsenic retention and remobilization in muddy sediments with high iron and sulfur contents from a heavily contaminated estuary in China[J]. Chemical Geology, 2012,314–317: 57-65
27 Yu Y M, Zhu Y X, Williams-Jones A E, et al. A kinetic study of the oxidation of arsenopyrite in acidic solutions: implications for the environment[J]. Applied Geochemistry, 2004,19(3): 435-444
28 Lengke M F, Tempel R N. Natural realgar and amorphous AsS oxidation kinetics[J]. Geochimica Et Cosmochimica Acta, 2003,67(5): 859-871
29 Long H, Dixon D G. Pressure oxidation kinetics of orpiment (As(2)S(3)) in sulfuric acid[J]. Hydrometallurgy, 2007,85(2-4): 95-102
30 Yu Y M, Zhu Y X, Gao Z M, et al. Rates of arsenopyrite oxidation by oxygen and Fe(III) at pH 1.8-12.6 and 15-45 degrees C[J]. Environmental Science & Technology, 2007,41(18): 6460-6464
XAFS study on oxidation dissolution of arsenopyrite in O2-bearing solution
ZHANG Guoqing1,2ZHANG Mingmei1WANG Shaofeng1JIA Yongfeng1
1(Key Laboratory of Pollution Ecology and Environmental Engineering,Institute of Applied Ecology,
Chinese Academy of Sciences,Shenyang 110016,China)
2(University of Chinese Academy of Sciences,Beijing 100049,China)
Background:One of the most important pollution sources of arsenic is the oxidation dissolution of arsenopyrite in oxygen-bearing solution.Purpose:This work investigated the oxidation process and product of arsenopyrite in O2-bearing solution using batch experiment.Methods:The influences of pH and dissolved oxygen (DO) on the oxidation dynamics of arsenopyrite are investigated. Besides that we also analyze the transformation of arsenic in the process of the oxidation of arsenopyrite using X-ray Absorption Fine Structure (XAFS).Results:XAFS spectroscopy data shows that, at pH=4, the oxidation of arsenopyrite is intensive and the released As(V) would be absorbed by iron oxide formed in the process of oxidation; at pH=7, the released As(III) would be absorbed by iron oxide and a few realgar analogues appear in the surface of arsenopyrite; at pH=9, the oxidation of arsenopyrite almost does not occur, but only a small amount of thioarsenates exhibit in the surface of arsenopyrite.Conclusion:The pH is the most important parameter controlling the oxidation of arsenopyrite. The oxidation of arsenopyrite is inhibited in neutral and alkaline conditions, due to the formation of Fe oxides on the surface of arsenopyrite quickly.
Arsenopyrite (FeAsS), Oxidation reaction, Iron oxides coatings, XAFS
ZHANG Guoqing, male, born in 1990, graduated from Henan Normal University in 2014, master student, focusing on environmental engineering
WANG Shaofeng, E-mail: wangshaofeng@iae.ac.cn
TL99
10.11889/j.0253-3219.2016.hjs.39.110103
国家自然科学基金(No.41530643、No.41473111)资助
张国庆,男,1990年出生,2014年毕业于河南师范大学,现为硕士研究生,研究领域为环境工程专业
王少锋,E-mail: wangshaofeng@iae.ac.cn
Supported by National Natural Science Foundation of China (No.41530643, No.41473111)
2016-08-16,
2016-10-11
