有机⁃无机杂多酸类离子液体催化汽油超声氧化脱硫
2016-12-14于凤丽谢盼辉朱国强解从霞于世涛
于凤丽,谢盼辉,朱国强,袁 冰,解从霞,于世涛
(1.青岛科技大学生态化工国家重点实验室培育基地,2.化工学院,青岛266042)
有机⁃无机杂多酸类离子液体催化汽油超声氧化脱硫
于凤丽1,谢盼辉1,朱国强1,袁冰1,解从霞1,于世涛2
(1.青岛科技大学生态化工国家重点实验室培育基地,2.化工学院,青岛266042)
合成了一系列有机⁃无机杂多酸类离子液体,并将其应用于超声作用下的催化模拟汽油氧化脱硫反应.结果表明,在超声波辅助下,不仅反应时间大大缩短,而且脱硫效率也大幅提高.在合成的一系列催化剂中,Zr0.25[BMIM]HPW12O40表现出最佳的催化活性.考察了超声波功率、超声/间隙时间、催化剂用量、H2O2用量、反应温度及反应时间等因素对脱硫效果的影响.以Zr0.25[BMIM]HPW12O40为催化剂,在优化的条件下[n(Cat.)=0.008 mmol,V(H2O2)=40 μL,V(模拟油)=10 mL,V(乙腈)=1 mL,温度25℃,时间10 min,超声功率300 W,超声时间2 s,间隙时间1.5 s],二苯并噻吩(DBT)的脱硫率达到97.8%;该催化剂循环使用5次后,脱硫率仍为81.9%;其对不同硫化物的催化活性顺序为DBT>4,6⁃二甲基苯并噻吩(4,6⁃DMDBT)>乙硫醚>苯硫醚>正丁硫醇>甲基苯基硫醚>苯并噻吩(BT)>噻吩.
杂多酸离子液体;氧化脱硫;萃取脱硫;超声;二苯并噻吩
随着全球范围内燃油使用量的不断增大,燃油燃烧后产生的废气对环境的危害也日趋严重.为此,世界各国对油品中的硫含量均制定了相当严格的标准.2008年,欧盟及美国等发达国家相继要求汽油和柴油中的硫含量低于10 μg/g[1].目前,我国只能实现将燃油中的硫含量降低到50 μg/g,远高于欧美国家的标准.因此,为满足环保要求和提高我国油品的国际竞争力,并使油品的硫含量全面与国际接轨,开展燃油深度脱硫已成为当务之急[2].
目前,燃油脱硫有多种方法,其中,催化加氢脱硫(HDS)技术在清洁燃油生产工艺中占主导地位.但HDS技术反应条件苛刻(高温、高压)、设备投资和操作费用高、氢气消耗量大,并且不能将燃油中的噻吩类硫化物完全脱除,故很难实现燃油的深度脱硫.因此,开发其它高效便捷的脱硫新工艺成为该领域研究的热点.催化氧化脱硫(ODS)因其反应条件温和、脱硫率高、操作工艺简单且绿色环保等优点,而备受研究者关注.ODS工艺的原理是:首先利用合适的氧化剂在催化剂作用下将油品中的硫化物氧化成亚砜和砜,再通过萃取、吸附和蒸馏等方法除去油品中的亚砜和砜.由于氧化产物亚砜和砜在硫原子上引入了氧原子,比相应的硫化物极性增强,因此更容易通过其它方法脱除,即更容易实现深度脱硫.目前,在ODS工艺中采用的氧化剂主要包括和O3[8,9]等.H2O2作为氧化剂的主要优点是易得、氧化条件温和、操作方便、副产物为水及无污染等.以H2O2为氧化剂的脱硫反应通常需要催化剂.近年来,杂多化合物被许多研究者用作氧化脱硫的催化剂[10~14],而有机⁃无机杂多化合物由于具有良好的两相相溶性,作为一类新型的绿色材料备受关注[15~17].Zhu等[18]分别用4种杂多酸离子液体[MIMPS]3PW12O40,[BMIM]3PW12O40,[BMIM]3PMo12O40和[BMIM]3SiW12O40作为催化剂,催化H2O2进行柴油深度脱硫反应.结果表明,[MIMPS]3PW12O40的催化性能最好,30℃下反应1 h脱硫率为100%.
将反应置于超声环境下,不仅能够缩短反应时间,而且可以提高反应效果[19,20].韩雪松等[19]在超声波作用下进行柴油深度氧化脱硫研究发现,在H2O2⁃有机酸催化体系中,脱硫率达到了94.8%,而未超声的脱硫率仅为67.2%,证实了超声反应的优越性.美国Sulphco公司的超声化学氧化脱硫项目取得了令人瞩目的成果,在中试实验中,短短20 min后有机硫化物的氧化转化率即可达到100%,大大缩短了反应时间,避免了H2O2的过度分解,并且改善了脱硫效果,中试所得产品的硫含量可以降低到约40 μg/g.
本文将具有氧化性的金属离子引入到杂多酸类离子液体中,合成了一系列含双氧化活性中心的杂多酸类离子液体,并将其用于超声作用下催化H2O2氧化模拟汽油脱硫.在室温下,仅需微量的催化剂催化反应10 min,模拟油品中的二苯并噻吩(DBT)即几乎全部氧化,说明合成的有机⁃无机杂多酸类离子液体具有极佳的催化性能.另外,此催化剂室温下为固态,反应后极易分离,且具有较佳的循环使用能力.
1 实验部分
1.1 试剂与仪器
磷钨酸(H3PW12O40)和正辛烷(C8H18)购自国药集团化学试剂有限公司;N⁃甲基咪唑(C4H6N2)、氯代正丁烷(n⁃C4H9Cl)、氯化锆(ZrCl4)、碳酸铈[Ce(CO3)3]、四氯化钛(TiCl4)、无水三氯化铁(FeCl3)、噻吩(C4H4S)、苯并噻吩(C8H6S)、二苯并噻吩(C12H8S)、4,6⁃二甲基苯并噻吩(C10H12S)、正丁硫醇(C4H10S)、乙硫醚(C4H10S)、苯硫醚(C12H10S)和甲基苯基硫醚(C7H8S)购自阿拉丁化学试剂有限公司;乙腈(C2H3N)购自烟台三和化学试剂有限公司;双氧水(H2O2,质量分数30%)购自上海科丰化学试剂有限公司.
Nicolet⁃510P型红外光谱仪(美国Nicolet公司);WK⁃2D型微库仑综合分析仪(江苏江分电分析仪器有限公司);Pogson⁃1000D型恒温密闭超声波反应器(南京普森仪器设备有限公司).
1.2 实验过程
1.2.1 催化剂的制备 有机⁃无机杂多酸类离子液体催化剂参照文献[21,22]的方法进行制备.以Zr0.25[BMIM]HPW12O40为例.1⁃丁基⁃3⁃甲基咪唑氯化盐([BMIM]Cl)参考文献[23]方法制备.称取2.332 g(1 mmol)ZrCl4和0.6986 g(4 mmol)[BMIM]Cl加入100 mL三口烧瓶中;取11.5202 g(4 mmol)磷钨酸置于烧杯中,加入40 mL去离子水使其溶解.将磷钨酸水溶液加入到三口烧瓶中,25℃下搅拌反应24 h.反应结束后,对反应液进行减压蒸馏除去大部分去离子水和少量盐酸,将剩余的溶液全部转移至坩埚中,于80℃真空干燥6 h,得到白色固体Zr0.25[BMIM]HPW12O40.其它催化剂的合成方法与上述方法类似.采用Nicolet⁃510P型红外光谱仪(KBr压片法)对催化剂进行红外表征,扫描范围为4500~400 cm-1.
1.2.2 模拟油的配制及脱硫实验 分别将一定量的4,6⁃二甲基苯并噻吩(4,6⁃DMDBT)、二苯并噻吩(DBT)、苯并噻吩(BT)、噻吩、甲基苯基硫醚、苯硫醚、乙硫醚和正丁硫醇溶于正辛烷中,配成一定浓度的模拟油品,以WK⁃2D型微库仑综合分析仪测定其硫含量.向超声反应器中依次加入定量催化剂、10 mL模拟汽油、定量H2O2(质量分数30%)和1 mL乙腈,在一定的超声功率、超声持续时间、超声间隙时间(前次超声终止和后次超声开始的时间间隔)和一定反应温度下,反应一定时间.反应结束后,将反应器中的反应液静置,移取上层油相至样品管中,测定其脱硫率.沉淀析出的催化剂经干燥后可重复使用.
2 结果与讨论
2.1 催化剂的红外光谱表征
在催化剂Zr0.25[BMIM]HPW12O40的红外谱图中,约1080,980,890和804 cm-1处出现Keggin结构的特征峰,分别归属于中心四面体和W—Ob2—W的伸缩振动吸收(其中Ob1和Ob2代表Keggin结构中2个不同的桥氧);3446 cm-1处的峰说明测试样品中可能含有少量水;3146 cm-1处的峰归属为咪唑环上的伸缩振动;2957 cm-1处的峰归属为咪唑环上烷基C—H的伸缩振动;1610和1562 cm-1处的峰分别归属为咪唑环上和双键的伸缩振动;1165 cm-1处的峰归属为烷基链C—C的伸缩振动.由此可见,该类催化剂中含有Keggin型杂多酸根和取代的咪唑环[BMIM]结构,与文献[24~27]报道一致.
2.2 不同催化剂的催化活性
在超声作用下,将合成的催化剂应用于模拟油品的氧化脱硫实验,结果如表1所示.由表1中En⁃try 1可知,单纯的Brönsted酸H3PW12O40因其氧化性较低,脱硫率仅有26.5%.当利用高价态的金属离子(Zr4+)部分或全部取代H3PW12O40中3个H+时,脱硫率得到显著提高(表1中Entries 2~4),这是因为高价态金属离子的引入增加了催化剂的氧化性.对比表1中Entries 3和4可知,具有一定Brönsted酸性的催化剂的催化效果更优.这是由于氧化剂H2O2在Brönsted酸性条件下更易被活化所致.为了增加催化剂与有机底物相的相溶性,利用有机阳离子取代磷钨酸酸式盐中的H+,合成了一系列有机⁃无机杂多酸离子液体.由表1中Entries 5和6可知,具有一定Brönsted酸性的催化剂的催化效果更优.由表1中Entries 6~10可知,含金属锆的催化剂Zr0.25[BMIM]HPW12O40呈现出最佳的催化活性(表1中Entry 6).

Table 1 Influence of different heteropolyacid catalysts on the desulfurization rate∗
在超声波辅助下,以Zr0.25[BMIM]HPW12O40为催化剂,在25℃下仅反应10 min,模拟油品的脱硫率即可达到94.2%;而无超声波作用时,在相同的催化剂和用量下,于40℃反应10 min,模拟油品的脱硫率仅为22.8%,延长反应时间至40 min时脱硫率达到90%,继续延长反应时间,脱硫率无明显提高.可见,超声辅助不仅可以提高催化效果,而且可以大大缩减反应时间.
2.3 最优实验条件的筛选
以Zr0.25[BMIM]HPW12O40为催化剂,考察了超声波功率、超声/间隙时间、催化剂用量、反应温度和反应时间等因素对模拟油品中DBT脱除效果的影响,筛选出最优实验条件.
2.3.1 超声功率对脱硫率的影响 在n(Cat.)=0.01 mmol,V(H2O2)=50 μL,V(模拟油)=10 mL,V(乙腈)=1 mL,温度25℃,时间10 min,超声时间和间隙时间都设定为1.5 s的条件下,考察了超声功率对催化H2O2氧化DBT脱硫的影响,结果如图1所示.由图1可见,DBT的脱硫率随着超声功率的增强而增大.当超声功率达到300 W时,脱硫率趋于稳定,继续增强超声功率,脱硫率升高的效果不明显.因此,选择300 W为超声脱硫的最佳功率.
2.3.2 超声时间对脱硫率的影响 在n(Cat.)=0.01 mmol,V(H2O2)=50 μL,V(模拟油)=10 mL,V(乙腈)=1 mL,温度25℃,时间10 min,超声功率300 W,超声间隙时间设定1.5 s的条件下,考察了超声时间对催化H2O2氧化DBT脱硫的影响,结果如图2所示.
由图2可见,DBT的脱硫率随着超声时间的延长,先是大幅增加,然后逐渐趋于平缓,说明超声时间在一定范围内对脱硫效果有较大影响.当超声时间达到2 s后,脱硫率随超声时间的延长基本不变.因此,选择2 s为脱硫的最佳超声时间.
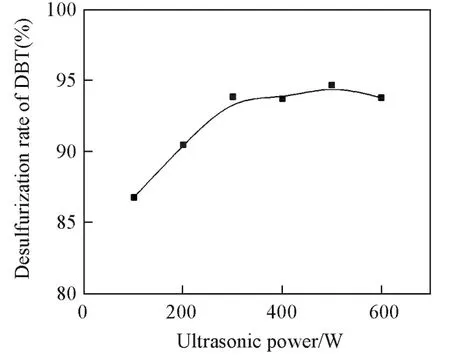
Fig.1 Influence of the ultrasonic power on the desulfurization rate

Fig.2 Influence of the ultrasonic time on the desulfurization rate
2.3.3 超声间隙时间对脱硫率的影响 在n(Cat.)=0.01 mmol,V(H2O2)=50 μL,V(模拟油)=10 mL,V(乙腈)=1 mL,温度25℃,时间10 min,超声功率300 W,超声时间设定2.0 s的条件下,考察了超声间隙时间对催化H2O2氧化DBT脱硫的影响,结果如图3所示.由图3可见,随着超声间隙时间的增长,DBT的脱硫率逐渐增加,当间隙时间达到1.5 s后脱硫率随超声间隙时间的延长基本不变.因此,选择1.5 s为最佳的超声间隙时间.
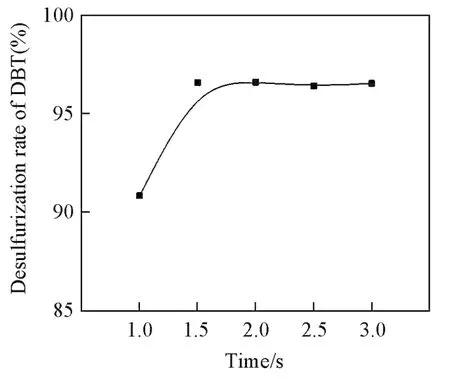
Fig.3 Influence of the ultrasonic off⁃time on the desulfurization rate

Fig.4 Influence of the amount of catalyst on the desulfurization rate
2.3.4 催化剂用量对脱硫率的影响 在V(H2O2)=50 μL,V(模拟油)=10 mL,V(乙腈)=1 mL,温度25℃,时间10 min,超声功率300 W,超声时间设定2 s,间隙时间设定1.5 s的条件下,考察了催化剂用量对催化H2O2氧化DBT脱硫的影响,其结果如图4所示.由图4可见,随着催化剂用量的增加,DBT的脱硫率逐渐增加,当催化剂用量达到0.008 mmol后,体系的脱硫率随着催化剂用量的增加基本保持不变.因此,最佳的催化剂用量定为0.008 mmol.
2.3.5 反应温度对脱硫率的影响 在n(Cat.)=0.008 mmol,V(H2O2)=50 μL,V(模拟油)=10 mL,V(乙腈)=1 mL,时间10 min,超声功率300 W,超声时间设定2 s,间隙时间设定1.5 s的条件下,考察了反应温度对催化H2O2氧化DBT脱硫的影响,其结果如图5所示.由图5可知,随着反应温度的升高,DBT的脱硫率先大幅度增加,后有所降低,当温度为25℃时,脱硫率达到最大值.随着温度的升高,脱硫率有所下降,这可能是由于H2O2分解速率加快,有效氧化活性降低所致.因此,选择25℃为超声脱硫的最佳反应温度.
2.3.6 反应时间对脱硫率的影响 在n(Cat.)=0.008 mmol,V(H2O2)=50 μL,V(模拟油)=10 mL,V(乙腈)=1 mL,反应温度25℃,超声功率300 W,超声时间设定2 s,间隙时间设定1.5 s的条件下,考察了反应时间对催化H2O2氧化DBT脱硫的影响,其结果如图6所示.由图6可见,随着反应时间的延长,DBT的脱硫率逐渐增加,当反应时间达到10 min后,脱硫率随着时间的延长基本保持不变,说明体系中的硫化物在10 min基本反应完毕.因此,选择10 min为超声脱硫的最佳反应时间.

Fig.5 Influence of the reaction temperature on the desulfurization rate
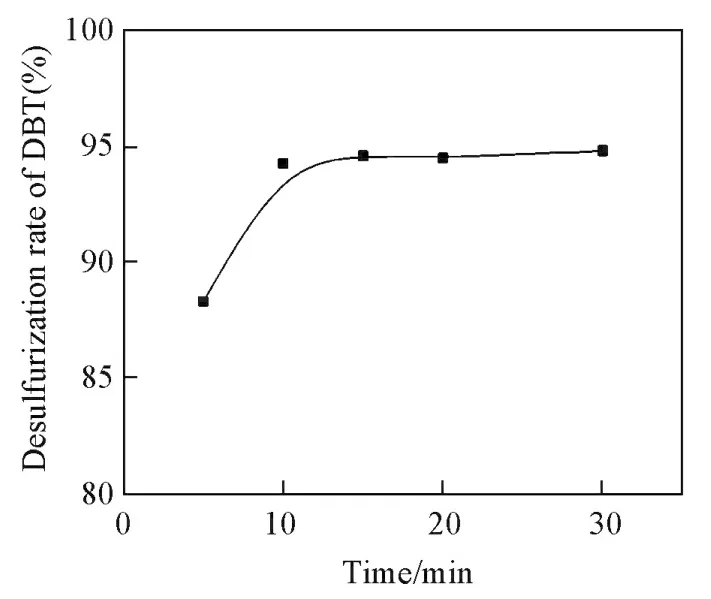
Fig.6 Influence of the reaction time on the desulfurization rate
2.3.7 H2O2用量对脱硫率的影响 在n(Cat.)=0.008 mmol,V(模拟油)=10 mL,V(乙腈)=1 mL,温度25℃,时间10 min,超声功率300 W,超声时间设定2 s,间隙时间设定1.5 s的条件下,考察了H2O2用量对催化氧化脱硫的影响,其结果如图7所示.由图7可见,随着H2O2用量的增加,DBT的脱硫率先上升后下降,当H2O2用量达到40 μL时,脱硫率达到最大值,继续增加H2O2用量,脱硫率反而下降.这可能是因为随着H2O2用量的增加,体系中水的含量增加,促使更多的杂多阴离子溶解,而催化剂阳离子部分倾向于进入油相,因此起主要催化作用的过氧磷钨酸通过水相“吸引作用”和阳离子的“排斥作用”,使催化活性中心与模拟油中硫化物的接触机会减少,从而降低了脱硫率.因此,选择40 μL为超声脱硫的最佳H2O2用量.

Fig.7 Influence of the amount of H2O2on the desulfurization rate
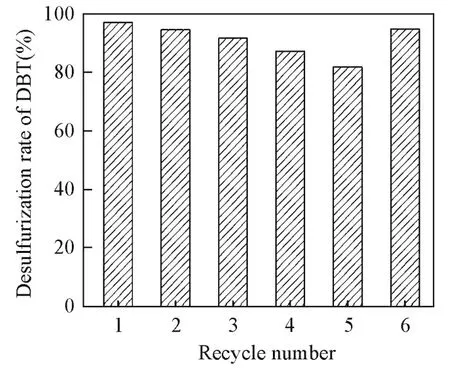
Fig.8 Influence of the recycle numbers of catalyst on the desulfurization rate
2.4 催化剂的循环使用寿命
在n(Cat.)=0.008 mmol,V(H2O2)=40 μL,V(模拟油)=10 mL,V(乙腈)=1 mL,温度25℃,时间10 min,超声功率300 W,超声时间设定2 s,间隙时间设定1.5 s的条件下,对催化剂的循环使用寿命进行了考察,其结果如图8所示.由图8可见,催化剂在循环使用过程中,脱硫率逐渐下降.催化剂循环使用5次,脱硫率由开始的97.8%降到81.9%.这主要是因为在每次反应结束后,催化剂都会出现少量的贴壁现象,致使下次进行脱硫实验时,催化剂的实际用量减少,使脱硫率下降.当补加由贴壁损失等量的催化剂后,脱硫率又得以恢复(循环使用6次).实验结果表明,该催化剂具有较佳的循环使用性能.
2.5 不同硫化物的脱除效果
以Zr0.25[BMIM]HPW12O40为催化剂,在最佳反应条件下,对模拟汽油中的不同硫化物进行脱硫实验,结果如表2所示.
由表2可知,催化剂Zr0.25[BMIM]HPW12O40对不同硫化物的脱除顺序为DBT>4,6⁃DMDBT>乙硫醚>苯硫醚>正丁硫醇>甲基苯基硫醚>BT>噻吩.硫化物的反应活性主要与它们的电子云密度及其空间位阻有关,噻吩类化合物的反应活性与文献[28]报道一致.

Table 2 Desulfurization efficiencies of different sulfides
3 结 论
制备了一系列含双氧化活性中心的有机⁃无机杂多酸类离子液体,并将其应用于催化模拟汽油的超声氧化脱硫反应.催化剂Zr0.25[BMIM]HPW12O40表现出最佳的催化活性,在最优反应条件[n(Cat.)=0.008 mmol,V(H2O2)=40 μL,V(模拟油)=10 mL,V(乙腈)=1 mL,温度25℃,时间10 min,超声功率300 W,超声时间2 s,间隙时间1.5 s]下,DBT的脱除率达到97.8%.催化剂Zr0.25[BMIM]HPW12O40也具有较佳的循环使用性能,循环使用5次后,脱硫率仍可达81.9%.该催化剂对不同硫化物的催化活性顺序为DBT>4,6⁃DMDBT>乙硫醚>苯硫醚>正丁硫醇>甲基苯基硫醚>BT>噻吩.
[1] Wu Y.,Xiao J.,Wu L.M.,Chen M.,Xi H.X.,Li Z.,Wang H.H.,J.Phys.Chem.C,2014,118(39),22533—22543
[2] Xiao J.,Bian G.,Zhang W.,Zhong L.,J.Chem.Eng.Data,2010,55(12),5818—5823
[3] Shiraishi Y.,Tachibana K.,Takayuki H.,Isao K.,Ind.Eng.Chem.Res.,2002,41(17),4362—4375
[4] Yu F.L.,Wang R.,Acta Chim.Sinica,2014,72(1),105—113(于凤丽,王睿.化学学报,2014,72(1),105—113)
[5] Liu Z.Q.,Xue F.,Lue Z.K.,Liu C.J.,Chem.J.Chinese Universities,2016,37(5),886—891(刘治庆,薛飞,雷振凯,刘晨江.高等学校化学学报,2016,37(5),886—891)
[6] Zhou X.,Li J.,Wang X.,Jin K.,Ma W.,Fuel Process.Technol.,2009,90(2),317—323
[7] Yu F.L.,Tang H.B.,Liu C.Y.,Xie C.X.,Yu S.T.,Acta Chim.Sinica,2014,72(11),1152—1156(于凤丽,唐会宝,柳春玉,解从霞,于世涛.化学学报,2014,72(11),1152—1156)
[8] Wang B.,Zhou J.P.,Ma H.Z.,J.Hazard.Mater.,2009,164(1),256—264
[9] Ma C.H.,Dai B.,Liu P.,Zhou N.,Shi A.J.,Ban L.L.,Chen H.W.,J.Ind.Eng.Chem.,2014,20(5),2769—2774
[10] Hao X.L.,Ma Y.Y.,Zang H.Y.,Wang Y.H.,Li Y.G.,Wang E.B.,Chem.Eur.J.,2015,21(9),3778—3784
[11] Zhang L.W.,Wu S.H.,Liu Y.,Wang F.F.,Han X.,Shang H.Y.,Appl.Organomet.Chem.,2016,30(8),684—690
[12] Zhang X.T.,Zhu Y.F.,Huang P.C.,Zhu M.Y.,RSC Adv.,2016,6,69357—69364
[13] Li Y.A.,Zhang M.,Zhu W.S.,Li M.,Xiong J.,Zhang Q.,Wei Y.C.,Li H.M.,RSC Adv.,2016,6,68922—68928
[14] Lu S.X.,Zhang H.,Wu D.,Han X.,Yao Y.,Zhang Q.Z.,RSC Adv.,2016,6,79520—79525
[15] Zhu T.,Lee Y.,Row K.,Chem.Res.Chinese Universities,2014,30(2),216—221
[16] Duan W.J.,Jiao S.H.,Liu X.,Chen J.L.,Cao X.,Chen Y.,Xu W.,Cui X.B.,Xu J.Q.,Pang G.S.,Chem.Res.Chinese Universi⁃ties,2015,31(2),179—186
[17] Chen J.Z.,Chen C.,Zhang R.,Guo L.,Hua L.,Chen A.J.,Xiu Y.H.,Liu X.R.,Hou Z.S.,RSC Adv.,2015,5,25904—25910
[18] Zhu W.,Huang W.,Li H.,Zhang M.,Jiang W.,Chen G.,Han C.,Fuel Process.Technol.,2011,92(10),1842—1848
[19] Han X.S.,Zhao X.S.,Dai Y.C.,Liu W.B.,Petroleum Process.Petrochem.,2006,37(2),30—33(韩雪松,赵德智,戴咏川,刘文豹.石油炼制与化工,2006,37(2),30—33)
[20] Liu L.,Lü H.,Xing J.J.,Qian J.H.,Sci.Technol.Eng.,2011,11(1),148—150(刘琳,吕宏,邢锦娟,钱建华.科学技术与工程. 2011,11(1),148—150)
[21] Yuan B.,Xie C.X.,Yu F.L.,Yang X.Y.,Yu S.T.,Zhang J.L.,Chen X.B.,SpringerPlus,2016,5,460—464
[22] Ramesh K.C.,Jagadeeswaraiah K.,Sai Prasad P.S.,Lingaiah N.,ChemCatChem,2012,4(9),1360—1367
[23] Chauvin Y.,Hirschauer A.,Olivier H.,J.Mol.Catal.,1994,92(2),155—165
[24] Xi Z.W.,Zhou N.,Sun Y.,Li K.L.,Science,2001,292(5519),1139—1141
[25] Zhou N.,Xi Z.W.,Cao G.Y.,Fan S.H.,J.Mol.Catal.(China),2001,15(2),113—118(周宁,奚祖威,曹国英,范淑华.分子催化,2001,15(2),113—118)
[26] Sun Y.,Xi Z.W.,Cao G.Y.,J.Mol.Catal.A:Chem.,2001,166(2),219—224
[27] Zhou N.,Xi Z.W.,Cao G.Y.,Gao S.,Appl.Catal.A:Gen.,2003,250(2),239—245
[28] Otsuki S.,Nonaka T.,Takashima N.,Qian W.,Ishihara A.,Imai T.,Kabe T.,Energy Fuels,2000,14(6),750—753
(Ed.:P,H,F,K)
†Supported by the National Natural Science Foundation of China(No.21476120),the Emphasis Development Plan of Shandong Province,China(No.2015GGX107008)and the Engineering Special Funding of Shandong Taishan Scholar,China(No.ts201511033).
Oxidative Desulfurization of Gasoline Catalyzed by Organic⁃inorganic Heteropoly Acid Ionic Liquids Under Ultrasound†
YU Fengli1,XIE Panhui1,ZHU Guoqiang1,YUAN Bing1,XIE Congxia1∗,YU Shitao2
(1.State Key Laboratory Base of Eco⁃chemical Engineering,2.College of Chemical Engineering,Qingdao University of Science and Technology,Qingdao 266042,China)
A series of Organic⁃inorganic heteropoly acid ionic liquids was synthesized and used for catalyzing oxidative desulfurization of simulated gasoline under ultrasound.With the help of ultrasonic wave,the reaction time was largely reduced,and the desulfurization efficiency was also raised.The results showed that Zr0.25[BMIM]HPW12O40exhibits the best catalytic activity.The effects of ultrasonic power,ultrasonic/clearance time,the amount of catalyst,reaction temperature,reaction time,and the amount of H2O2on the desulfurization rate were fully investigated.The selected optimal conditions were as follows:n(Cat.)=0.008 mmol,V(H2O2)=40 μL,V(simulated oil)=10 mL,V(acetonitrile)=1 mL,reaction temperature 25℃,reaction time 10 min,the ultrasonic power 300 W,the ultrasonic time 2 s,and the ultrasonic off⁃time 1. 5 s.Under the optimal conditions,the sulfur removal of DBT could reach 97.8%.The solid catalyst Zr0.25[BMIM]HPW12O40could be directly separated out after the reaction,and could be reused after the vacuum drying.The results showed that Zr0.25[BMIM]HPW12O40also exhibited good recyclability.After 5 recycles,the desulfurization rate still could reach 81.9%.By using Zr0.25[BMIM]HPW12O40as catalyst,the reaction activity decreased in the order of DBT>4,6⁃DMDBT>ethyl thioether>phenyl thioether>n⁃butyl mercaptan>methyl phenyl thioether>BT>thiophene.
Heteropoly acid ionic liquid;Oxidative desulfurization;Extractive desulfurization;Ultrasound;Dibenzothiophene
O643.3;O621
A
10.7503/cjcu20160539
2016⁃07⁃25.网络出版日期:2016⁃11⁃18.
国家自然科学基金(批准号:21476120)、山东省重点发展计划项目(批准号:2015GGX107008)和山东省泰山学者工程专项经费(批准号:ts201511033)资助.
联系人简介:解从霞,女,博士,教授,博士生导师,主要从事环境友好催化和生物质转化利用方面的研究.
E⁃mail:xiecongxia@126.com
