SCN1A基因突变阳性的Dravet综合征患儿的临床特征分析
2016-12-08黄绍平宋婷婷杨长虹王雪莹
李 丹,黄绍平,宋婷婷,杨 琳,杨长虹,王雪莹
(西安交通大学第二附属医院儿科,陕西西安 710004)
◇临床研究◇
SCN1A基因突变阳性的Dravet综合征患儿的临床特征分析
李 丹,黄绍平,宋婷婷,杨 琳,杨长虹,王雪莹
(西安交通大学第二附属医院儿科,陕西西安 710004)
目的 分析携带SCN1A基因突变的Dravet综合征患儿的临床表现特征,以期为临床治疗做出正确决策提供帮助。方法 回顾性分析了29名Dravet综合征患儿的临床资料(男性16名,女性13名,平均年龄3.2岁),并对患儿进行了SCN1A的基因检测、认知功能评价及脑电图分析。结果 19例患儿检测到SCN1A基因突变,包括错义突变、无义突变、剪接突变以及大片段缺失。SCN1A+组的平均发病年龄为6.07月,SCN1A-组为10.00月(t=3.465,P=0.020)。SCN1A+组首次发作即为癫痫持续状态(SE)的患儿比例为68.4%(13/19),SCN1A-组为10%(P<0.05),1岁以前月发作频率SCN1A+组为(5.44±1.62)次,SCN1A-组为(0.98±0.4)次(P<0.05)。SCN1A+组患儿脑电背景波出现弥漫性慢波的比例为73.7%(14/19),SCN1A-组为3例(30%)(P<0.05)。精神运动发育评估的结果在两组间无统计学差异(P>0.05)。结论 SCN1A基因突变阳性患儿发病年龄早、发作频率高、SE患儿比例高、脑电图背景更容易出现弥漫性慢活动。
癫痫;SCN1A基因;突变;Dravet综合征
目前已知的与Dravet综合征有关的基因突变位点有SCN1A、PCDH19、SCN2A、SCN1B等。SCN1A和PCDH19基因是目前突变发生频率最高的两个等位基因。Dravet综合征患儿中60%~80%携带有SCN1A基因突变[1-3]。SCN1A基因编码了神经元钠离子通道Nav 1.1的α1亚单位,它主要定位于染色体2q24.3位置。钠离子通道的变化可能导致神经元兴奋性的增加以及神经元出现同步化的放电,最终导致惊厥发作。一般来说,Dravet综合征患儿应避免选用钠离子通道阻滞剂,但SONEIJEN-SCHOUWENAARS[4]的一项研究表明,卡马西平或奥卡西平不一定都使得发作恶化,相反,对于发作有一定的控制作用,但如果患儿携带SCN1A基因突变应该避免使用卡马西平、奥卡西平等钠离子通道阻滞剂。故从临床上尽早识别Dravet综合征患儿是否可能携带SCN1A突变显得非常重要。本文旨在通过对携带SCN1A基因突变的Dravet综合征患儿的早期临床特征、脑电图表现以及生长发育情况进行分析,帮助临床医生尽早识别、尽早行基因检查确诊,以避免临床错误的选药。
1 对象与方法
1.1 研究对象 所有患儿均来自西安交通大学第二附属医院儿内科。纳入标准根据国际抗癫痫联盟对于Dravet综合征的诊断标准:①惊厥发作前发育正常;②1岁前出现热性或无热性惊厥,有家族史倾向;③逐渐发展为药物难治性惊厥;④发病前智力运动发育正常;⑤2岁以后逐渐出现精神运动发育停滞或倒退;⑥病初脑电图正常,随后逐渐出现多灶性棘慢波或多棘慢波发放。排除标准:癫痫发作前即出现精神运动发育迟缓患儿;发作类型中存在强直发作或失张力发作;虽然临床表现符合上述特点,但未行基因检测者;围产期出现严重的缺氧缺血病史、低血糖、脑梗塞、颅内感染或先天性脑发育畸形、遗传代谢病及其他的退行性病变等明确的神经系统损害者。共29例患儿纳入研究,其中男性16例,女性13例。
1.2 研究方法 收集和分析29例患儿的临床资料。癫痫持续状态(status epilepticus, SE)的诊断标准为惊厥发作或发作间期意识不清醒持续超过30 min以上。根据患儿的年龄及配合程度选择Geseel、Wechsler量表或临床观察评价所有患儿的认知情况。根据每位患儿详细的每日行为能力评定将发育评估结果分为正常(比如与其年龄相匹配的生活能力及正常的学校表现)、轻度智力低下(如因智力残疾造成的学校表现较差)、中度智力低下(如语言认知发育迟缓,但生活能自理)以及重度智力低下(生活不能自理)[5]。29例患儿行头颅磁共振检查。
1.3 基因分析 在研究对象监护人签署知情同意书后,采集研究对象的外周血5 mL,应用过柱法(QIAamp Blood DNA Mini Kit, QIAGEN公司,美国)提取外周血DNA,应用Primer Premier 5.0软件针对SCN1A基因的26个外显子编码区设计引物,应用PCR MasterMix聚合酶(TIANGEN,天根)进行PCR扩增(ABI9700型PCR仪,Life technology,美国),然后对PCR产物进行测序(ABI3500测序仪,Life technology,美国),与参考序列(NM_001165963.1与NG_011906.1)进行比较,从而发现可能存在的基因突变。基因突变阳性的患儿再对其父母进行一代测序的验证。对于SNC1A基因突变阴性的患儿进一步采用多重连接探针扩增技术(multiplex ligation-dependent probe amplification, MLPA)筛查SCNlA基因的片段缺失和重复。
1.4 数据分析 运用SPSS 16.0统计软件进行数据处理。计量资料用均数±标准差表示,数据在满足正态性和方差齐性的条件下,采用t检验。计数资料的比较采用卡方检验或Fisher确切概率法。P≤0.05为差异有统计学意义。
2 结 果
2.1 临床资料 29例患儿最后一次随访时平均年龄3.3岁((IQR 1.50~4.25岁,1.2~7.7岁),平均随访时间2.1年(4月~4.6年)。惊厥首次发作的平均年龄是7.6月。SE的发生率为75.9%(22/29),惊厥发作时间最长的1例患儿超过5 h。10例患儿首次惊厥发作即为SE。3例患儿的一级亲属被诊断为癫痫。26例患儿首次发作由发热诱发,最低触发温度为37.8 ℃。27例患儿(93.1%)有两种或更多形式的发作类型,包括全面性强直阵挛发作、全面性阵挛、半侧阵挛、部分性发作、肌阵挛发作等。27例(93.1%)患儿使用了超过两种的抗癫痫药物。
2.2 SCN1基因分析结果 基因测序分析结果显示,共19例(65.6%)患儿SCN1基因突变为阳性,共检测到了18个突变位点及1例SCN1A基因大片段缺失,突变类型包括15例错义突变,2例移码突变及1例剪接突变。行父母SCN1A基因验证的有16例,14例(87.5%)为新发突变,2例来源于母亲,其中1例母亲为体细胞嵌合体,另1例母亲为杂合体,且有癫痫病史(表1)。图1显示25号患儿及父母基因突变及父母亲验证情况。SCN1A基因一代测序显示患儿为杂合子,父亲基因检测正常,母亲曾经有过抽搐发作,但未携带SCN1A突变基因。
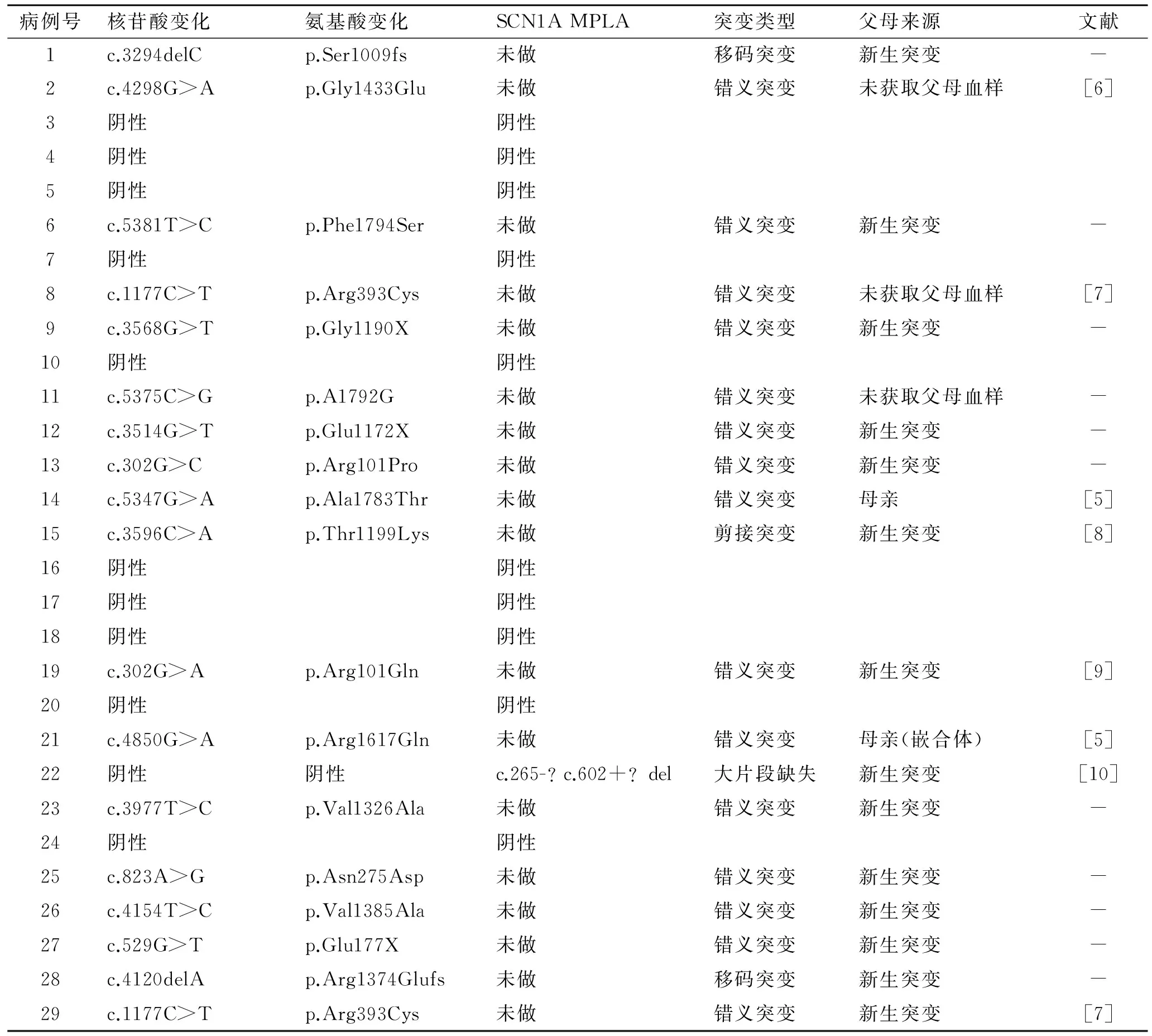
表1 Dravet综合征患儿SCN1A基因突变Tab.1 SCN1A gene mutation of Dravet syndrome children
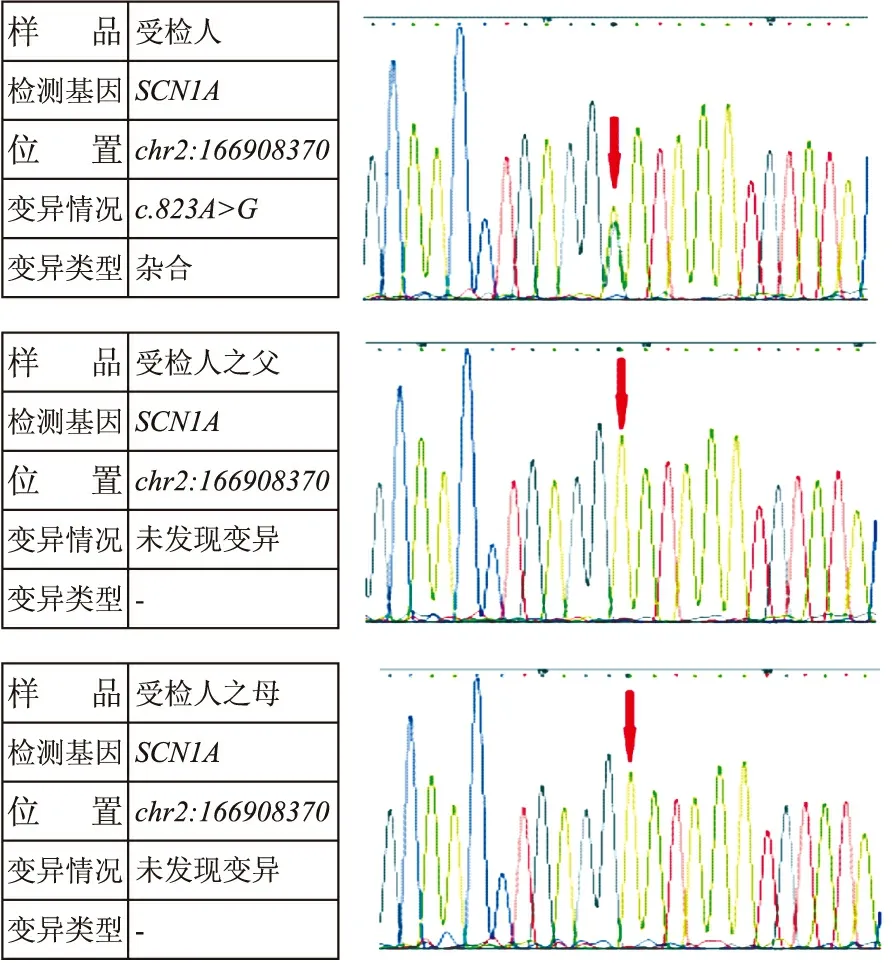
图1 25号患儿基因检测显示c.823A>G基因突变,其父母SCN1A基因未发现变异。患儿母亲惊厥2次,但未携带SCN1A突变基因。Fig.1 Family study of patient 25 with novel SCN1A mutations. c.823A>G (p.Asn275Asp) mutation was found in patient 25. Genetic testing of his parents was normal. His mother had undergone two convulsions; however, she did not carry any mutation in SCN1A.
2.3 SCN1A+和SCN1A-两组患儿临床指标的对比 依据SCN1A的基因检测结果将29例患儿分成SCN1A+组和SCN1A-组,两组患儿最后一次随访时的年龄相比无统计学差异。SCN1A+的平均发病年龄为6.07月,小于SCN1A-组。SCN1A+组首次发作即为SE的患儿比例为68.4% (13/19),而SCN1A-组为10%。1岁以前月发作频率SCN1A+组为(5.44±1.62)次,SCN1A-组为(0.98±0.4)次。其余临床数据两组之间比较无统计学差异(表2)。
29例患儿脑电图均描记出发作间期的癫痫波发放。7例(24.1%)患儿描记到多棘慢波,局灶性放电的患儿比例(14例,48.3%)接近全导放电(15例,51.7%)的比例。9例患儿(31.3%)出现睡眠结构紊乱。SCN1A+组患儿脑电背景波出现弥漫性慢波的比例为73.7%(14/19),SCN1A-组为3例(30%),两组具有统计学差异(Fisher确切概率法,P=0.035)。两组在多棘慢波患儿比例、发作间期癫痫波放电模式以及有无睡眠结构紊乱方面均无统计学差异。
29例患儿中有10例(34.5%)精神运动发育正常,10例(34.5%)轻度精神运动发育迟缓,6例(20.7%)中度精神运动发育迟缓,3例(10.4%)重度精神运动发育迟缓。SCN1A+和SCN1A-两组在正常、轻度、中度及重度认知损害方面均无统计学差异(表2)。
2.4 影像学检查结果 除了4例患儿外,其余患儿均进行了头颅磁共振检查。患儿的磁共振检查基本正常,或表现为非特异性异常,包括松果体囊肿及基底节脑软化灶。
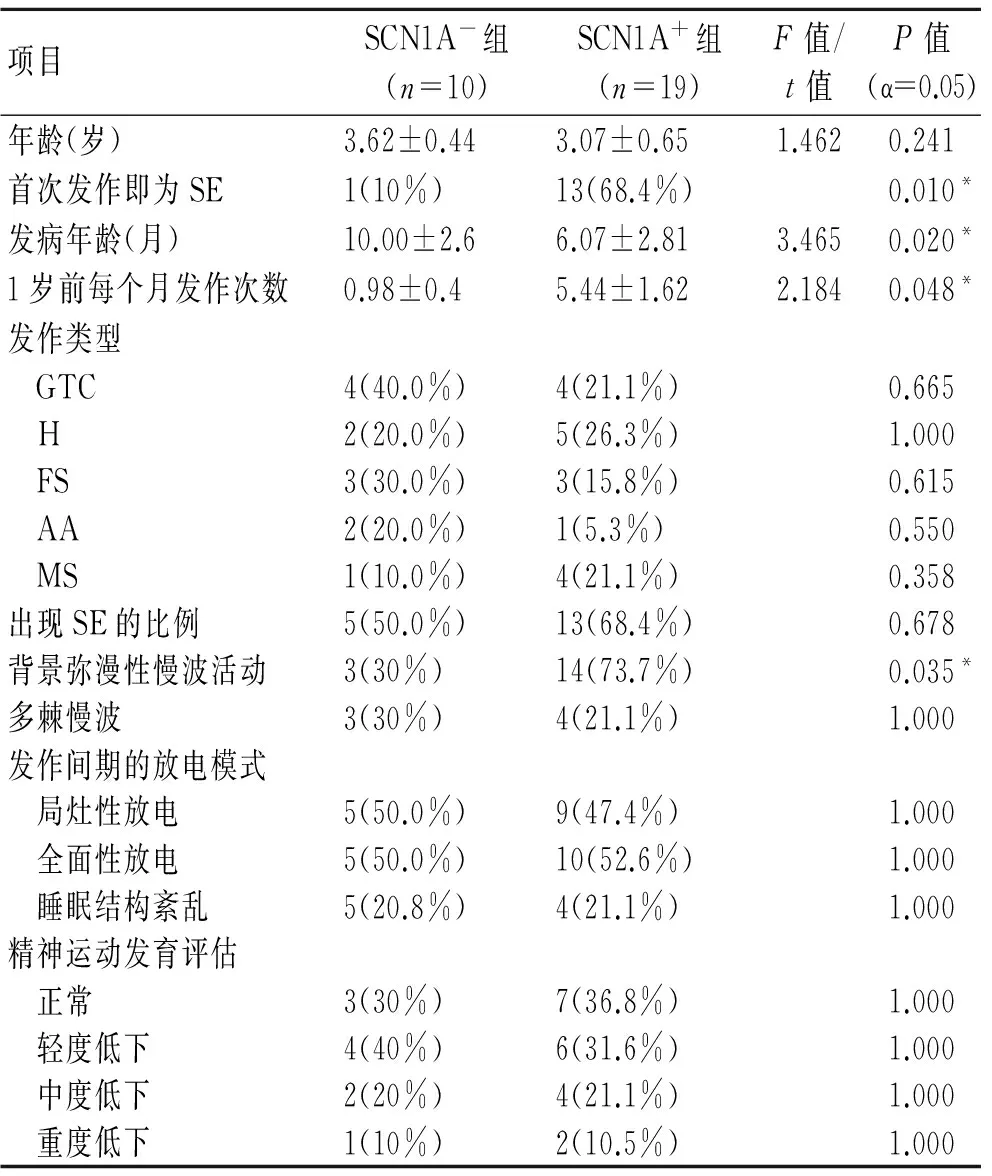
表2 SCN1A+和SCN1A-两组患儿临床特征的比较Tab.2 Comparison of clinical features in SCN1A+and SCN1A-groups
3 讨 论
流行病学资料显示Dravet综合征的发病率在1∶20 000和1∶40 000之间,男女比例为2∶1[11]。其临床特征为①首次发作通常为发热诱发的持续较长时间的惊厥发作,可以是全面性或部分性发作;②逐渐发展成为多种发作形式,如肌阵挛发作、部分性发作、非典型性失神、失张力发作等;③大多数患儿从生后的第2年逐渐出现不同程度精神运动发育的停滞或倒退[12]。
本研究中,19例DS患儿检测到18个SCN1A基因位点的突变,错义突变(16/19,84.2%)是SCN1A基因出现频率最高的突变类型。以往的文献报道SCN1A的突变率为40%~80%。PETRELLI等[13]的研究表明在Dravet综合征患儿中有75%的患儿SCNA1基因突变阳性。一项大样本的研究表明448例Dravet综合征患儿中有188名患儿(42%)检测到SCN1A基因的192个基因位点的突变,其中错义突变所占的比例为47.45%[3]。来自于台湾的一项基因研究表明SCN1A基因突变阳性的Dravet综合征患儿比例为55.6%[14]。这些研究结果差异的原因可能和样本的大小以及诊断标准的不同有关。
本研究表明,SCN1A+组患儿的1岁以前的月发作次数、首次发作即为SE的患儿比例明显高于SCN1A-组,并且发病年龄更早,与RAGONA等[1]研究结果一致。PETRELLI等[13]证实SCN1A+患儿在1岁以前具有更高频率的惊厥发作。LE GAL等[14]的数据表明SCN1A突变阳性患儿发生首次SE的年龄为8个月。最开始的惊厥表现为反复的全面性或半侧阵挛发作。发作通常持续时间较长,或在1 d 内成簇发作,甚至发生SE[15]。SCN1A和PCDH19基因是目前突变发生频率最高的两个等位基因,PCDH19基因是PCDH19,即原钙黏蛋白19,是一种跨膜蛋白,属于钙依赖的细胞粘附分子家族中的一员,位于X染色体长臂2区2带[16]。其致病机制和SCN1A不同,故此基因突变导致的Dravet综合征可以使用钠离子通道阻滞剂。通过对SCN1A突变阳性的Dravet综合征患儿临床特征的分析,将有助于临床医生尽早识别,正确的选择抗癫痫药物。
既往鲜有文献报道SCN1A+和SCN1A-组患儿的发作间期脑电图特点。本研究结果表明,SCN1A+组患儿出现背景弥漫性慢波的比例是73.7%,超过SCN1A-组患儿。最近的研究表明,SCN1A基因突变阳性的Dravet综合征患儿随着年龄的增长更容易出现前头部慢波活动或弥漫性背景慢波活动[17]。脑电图出现弥漫性慢波的原因可能归因于SCN1A基因的突变。研究发现SCN1A E1099X/+(Het)大鼠的齿状回颗粒细胞形态异常,而且颗粒细胞的树突生长减少,棘突生长增多,这一改变可能会影响神经元网络活性[18],可能使得脑功能弥漫性受损,出现弥漫性背景慢波活动。尽管关于二者之间直接的联系尚未见文献报道,但是多种因素包括SCN1A基因突变导致的神经元钠离子通道失活可能是造成患儿脑电图恶化的主要原因。
患儿生长发育在发病的早期是正常的,在出生后第2年出现迟滞甚至倒退。一些研究发现惊厥发作的频次[19]或肌阵挛发作[20],或SE[1]的存在可能与预后不良有关。RAGONA等[1]使用韦氏智力测评量表发现6岁以后的Dravet综合征患儿无智商正常者。这种智力发育的倒退或停滞归因于SCN1A基因突变还是多种因素作用的结果尚无定论。本研究发现,智力正常、轻度受损、中度受损或重度受损的患儿比例在SCN1A基因突变阳性组和阴性组中均无统计学差异。这表明SCN1A基因突变可能不是造成认知受损的主要原因。既往的文献报道对此也存在争议。RAGONA等[1]指出药物治疗的作用可能是造成认知损害的原因之一,因为年长一些的患儿可能使用了更多的药物或更大的剂量。与之相反的是VILLENEUVE[21]的队列研究发现认知落后与多药治疗无关,因为单药治疗的Dravet综合征患儿并没有表现出比多药治疗的患儿更好的认知水平。动物实验中SCN1A基因敲除大鼠在没有发作情况下,也似乎表现出了认知受损[22]。或许多种因素比如频繁发作,多药治疗及不合理的使用抗癫痫药物、SCN1A基因的突变都将导致认知发育的落后或停滞。
本项研究表明SCN1A突变阳性组患儿临床特征为:癫痫发作出现的更早,每月发作频率高,首次发作即为SE的患儿比例更高,以及脑电图显示弥漫性慢活动的比例更高。了解这些特征,将有助于临床医生尽早识别出患儿是否携带SCN1A基因突变,尽早行基因检查确诊,为临床治疗做出正确决策提供帮助。
[1] RAGONA F, BRAZZO D, DE GIORGI I, et al. Dravet syndrome: early clinical manifestations and cognitive outcome in 37 Italian patients[J]. Brain Dev, 2010, 32(1):71-77.
[2] MORSE RP. Dravet syndrome: inroads into understanding epileptic encephalopathies[J]. J Pediatr, 2011,158 (3):354-359.
[3] WANG JW, SHI XY, KURAHASHI H, et al. Prevalence of SCN1A mutations in children with suspected Dravet syndrome and intractable childhood epilepsy[J]. Epilepsy Res, 2012,102 (3):195-200.
[4] SONEIJEN-SCHOUWENAARS FM, VEENDRICK MJ, VAN MIERLO P, et al. Carbamazepine and oxcarbazepine in adult patients with Dravet syndrome: Friend or foe? [J]. Seizure, 2015, 29(3):114-118.
[5] HARKIN LA, MCMAHON JM, LONA X, et al. The spectrum of SCN1A-related infantile epileptic encephalopathies[J]. Brain, 2007, 130(Pt 3):843-52.
[6] SUN H, ZHANG Y, LIANG J, et al. Seven novel SCN1A mutations in Chinese patients with severe myoclonic epilepsy of infancy[J]. Epilepsia, 2008, 49(6):1104-1107.
[7] LIM BC, HWANG H, CHAE JH, et al. SCN1A mutational analysis in Korean patients with Dravet syndrome[J]. Seizure, 2011, 20 (10):789-794.
[8] SUN H, ZHANG Y, LIU X, et al. Analysis of SCN1A mutation and parental origin in patients with Dravet syndrome[J]. J Hum Genet, 2010, 55 (7):421-427.
[9] FUKUMA G, OGUNI H, SHIRASAKA Y, et al. Mutations of neuronal voltage-gated Na+channel alpha 1 subunit gene SCN1A in core severe myoclonic epilepsy in infancy (SMEI) and in borderline SMEI (SMEB)[J]. Epilepsia, 2004, 45(2):140-148.
[10] GUERRINI R, CELLINI E, MEI D, et al. Variable epilepsy phenotypes associated with a familial intragenic deletion of the SCN1A gene[J]. Epilepsia, 2010, 51(12):2474-2477.
[11] CLAES L, CEULEMANS B, AUDENAERT D, et al. De novo SCN1A mutations are a major cause of severe myoclonic epilepsy of infancy[J]. Hum Mutat, 2003, 21(6):615-621.
[12] DRAVET C, BUREAU M, OGUNI H, et al. Severe myoclonic epilepsy in infancy: Dravet syndrome [J]. Adv Neurol, 2005, 95:71-102.
[13] PETRELLI C, PASSAMONTI C, CESARONI E, et al. Early clinical features in Dravet syndrome patients with and without SCN1A mutations [J]. Epilepsy Res, 2012, 99(1-2):21-27.
[14] LE GAL F, LEBON S, RAMELLI GP, et al. When is a child with status epilepticus likely to have Dravet syndrome?[J]. Epilepsy Res, 2014, 108 (4):740-747.
[15] GUERRINI R. Dravet syndrome: the main issues[J]. Eur J Paediatr Neurol, 2012, 16 (Suppl 1):S1-4.
[16] MONICA G, GRAZIA A, MICHELA S, et al. PCDH19 mutation in female patients from southern Italy[J]. Seizure, 2015, 24:118-120.
[17] DEPIENE C, BOUTEILLER D, KEREN B, et al. Sporadic infantile epileptic encephalopathy caused by mutations in PCDH19 resembles Dravet syndrome but mainly affects females[J]. PLoS Gene, 2009, 5(2):e1000381.
[18] TSAI MS, LEE ML, CHANG CY, et al. Functional and structural deficits of the dentate gyrus network coincide with emerging spontaneous seizures in a Scn1a mutant Dravet Syndrome model during development [J]. Neurobio Dis, 2015, 77:35-48.
[19] KIM SH, NORDI DR, BERG AT, et al. Ictal ontogeny in Dravet syndrome[J]. Clin Neurophys, 2015, 126(3):446-455.
[20] WOLFF M, CASSE PC, DRAVET C. Severe myoclonic epilepsy of infants (Dravet syndrome): natural history and neuropsychological findings[J]. Epilepsia, 2006, 47 (Suppl 2):45-48.
[21] VILLENEUVE N, LAGUITTON V, VIELLARD M, et al. Cognitive and adaptive evaluation of 21 consecutive patients with Dravet syndrome[J]. Epilepsy Beha, 2014, 31:143-148.
[22] BATTAGLIA D, CHIEFFO D, SIRACUSANO R, et al. Cognitive decline in Dravet syndrome: is there a cerebellar role?[J]. Epilepsy Res, 2013, 106(1-2):211-221.
(编辑 邱 芬)
Clinical features of Dravet syndrome patients with SCN1A gene mutations
LI Dan, HUANG Shao-ping, SONG Ting-ting, YANG Lin,YANG Chang-hong, WANG Xue-ying
(Department of Pediatrics, the Second Affiliated Hospital of Xi’an Jiaotong University, Xi’an 710004, China)
Objective To analyze the characteristics of the clinical manifestations of Dravet syndrome (DS) children with SCN1A gene mutations so as to help pediatricians to make right decisions to treat DS patients. Methods Twenty-nine DS patients (16 males and 13 females, mean age of 3.2 years old) were retrospectively studied. The clinical records, including SCN1A gene test, psychomotor developmental assessmentsm, and electroencephalogram, were collected. Results SCN1A gene mutation was detected in 19 cases, including missense mutation, nonsense mutation, splicing mutation and a deletion. The average age of seizure onset was 6.07 months in SCN1A+group, 10.00 months in SCN1A-group (t=3.465,P=3.465). The ratio of first seizure as epilepticus status was 68.4% (13/19) in SCN1A+group and 10% in SCN1A-group. The attacks per month before the age of 1 year was 5.44±1.62 times in SCN1A+group and 0.98±0.4 times in SCN1A-group. The ratio of diffuse slowing in the ictal electroencephalogram was 73.7% (14/19) in SCN1A+group. All the above indexes were significantly different. Psychomotor developmental assessment results did not differ between the two groups (P>0.05). Conclusion DS patients in SCN1A mutation positive group manifested early onset of seizures, long duration of the first seizure, frequent episodes before the first year and diffuse slow activity in electroencephalograph.
epilepsy; SCN1A gene; mutation; Dravet syndrome
2016-01-20
2016-06-29
西安交通大学第二附属医院科研基金青年资助项目[No.YJ(QN)201213]Supported by the Youth Fund Scientific Research Project of the Second Affiliated Hospital of Xi’an Jiaotong University [No.YJ(QN)201213]
李丹. E-mail: runningtortoise@163.com
R748
A
10.7652/jdyxb201606014
优先出版:http://www.cnki.net/kcms/detail/61.1399.R.20161012.1600.012.html(2016-10-12)
