Cardiovascular dysfunction following spinal cord injury
2016-12-02ElizabethPartidaEugeneMironetsShaopingHouVeronicaTom
Elizabeth Partida, Eugene Mironets, Shaoping Hou, Veronica J. Tom
Spinal Cord Research Center, Department of Neurobiology & Anatomy, Drexel University College of Medicine, Philadelphia, PA, USA
INVITED REVIEW
Cardiovascular dysfunction following spinal cord injury
Elizabeth Partida, Eugene Mironets, Shaoping Hou, Veronica J. Tom*
Spinal Cord Research Center, Department of Neurobiology & Anatomy, Drexel University College of Medicine, Philadelphia, PA, USA
Both sensorimotor and autonomic dysfunctions often occur after spinal cord injury (SCI). Particularly, a high thoracic or cervical SCI interrupts supraspinal vasomotor pathways and results in disordered hemodynamics due to deregulated sympathetic outflow. As a result of the reduced sympathetic activity, patients with SCI may experience hypotension, cardiac dysrhythmias, and hypothermia post-injury. In the chronic phase, changes within the CNS and blood vessels lead to orthostatic hypotension and life-threatening autonomic dysreflexia (AD). AD is characterized by an episodic, massive sympathetic discharge that causes severe hypertension associated with bradycardia. The syndrome is often triggered by unpleasant visceral or sensory stimuli below the injury level. Currently the only treatments are palliative - once a stimulus elicits AD, pharmacological vasodilators are administered to help reduce the spike in arterial blood pressure. However, a more effective means would be to mitigate AD development by attenuating contributing mechanisms, such as the reorganization of intraspinal circuits below the level of injury. A better understanding of the neuropathophysiology underlying cardiovascular dysfunction after SCI is essential to better develop novel therapeutic approaches to restore hemodynamic performance.
blood pressure; heart rate; autonomic dysreflexia; hypertension; bradycardia; spinal cord lesion; sprouting; plasticity; bladder distension; relay; sympathetic activity
Introduction
Traumatic spinal cord injury (SCI) interrupts connections between higher centers and the spinal cord and compromises body activity. Both sensorimotor deficits and autonomic disorders greatly influence the functional, psychological, and socioeconomic aspects of patients’ lives. Compared to numerous investigations on motor and sensory paralysis, however, autonomic dysfunctions have not been the focus of basic or clinical research for a long time. In recent decades autonomic disorders after SCI have drawn more investigations as researchers and clinicians began to emphasize their clinical priority. The disruption of descending autonomic pathways renders abnormalities in multiple organ systems including cardiovascular function, respiration, gastrointestinal function, micturition, sexual function, sudomotor activity, and thermoregulation. Among various complications that individuals with SCI experience, cardiovascular dysfunction often develops when the injury occurs at the cervical or high thoracic level, resulting in the loss of supraspinal regulation of spinal sympathetic activity. In particular, life-threatening autonomic dysreflexia (AD), which manifests as a sudden episodic hypertension and reflexic bradycardia, can be triggered by unpleasant stimuli below the injury level. Latest epidemiological studies revealed that hundreds of new cases occur each year globally (Lee et al., 2014). Notably, disordered hemodynamics is one of the leading causes of morbidity and mortality in SCI patients (Garshick et al., 2005). Here, we will provide an overview of how the nervous system controls cardiovascular function normally and how modulation of this system is altered following a severe spinal cord injury because of the loss of supraspinal innervation. Additionally, we will discuss how injury-induced plasticity caudal to the injury is another factor that contributes to the development of AD.
Neural Control of Cardiovascular Function
The neural circuitry controlling cardiovascular function is comprised of multiple populations of neurons in the central and peripheral nervous system. Arterial chemoreceptors and cardiopulmonary baroreceptors convey excitatory primary afferent inputs to the nucleus tractus solitarius in the brainstem via the vagus and glossopharyngeal cranial nerves. Baroreceptors in the aortic arch, carotid sinus, and coronary arteries detect changes in arterial pressure whereas chemoreceptors in the carotid bodies respond to the changes in partial pressure of oxygen and carbon dioxide in the blood (Loewy, 1990). The nucleus of the solitary tract is a major recipient of primary sensory cardiovascular information, which, in turn, precisely regulates hemodynamic performance.
Successful cardiovascular regulation is achieved through the exchange of counteracting responses between the two divisions of the autonomic nervous system: the sympathetic and parasympathetic nervous systems. In normal conditions, sympathetic excitation mediates an increase in heart rate (HR) and blood pressure (BP) while parasympathetic activation elicits a reduction of heart rate and reflexive decrease of blood pressure. Sympathetic preganglionic neurons (SPNs) are mainly situated within the intermediolateral cell column (IML) in the lateral horn and gray commissure around the central canal at the T1-L2spinal cord levels (Figure 1A, B; orange neurons).Some SPNs are also present in the nucleus intercalatus (the gray matter between IML and central canal). The heart is innervated by both sympathetic and parasympathetic systems whereas most peripheral blood vessels receive only sympathetic input. SPNs related to cardiac regulation are located at T1-4spinal cord levels. These neurons send extensions to postganglionic neurons in the superior, middle cervical, and stellate ganglia (Figure 1A, B; green neurons). The fibers of the postganglionic neurons in these ganglia, in turn, project and terminate in the target organ (i.e., the heart and the peripheral vessels). The parasympathetic preganglionic neurons (PPNs) innervating the heart are situated in the dorsal motor nucleus of the vagus (DMV) and the nucleus ambiguous in the medulla oblongata. Their projections extend via the vagus and glossopharyngeal nerves to the postganglionic neurons, which are present within the epicardium or the cardiac walls adjacent to the sinoatrial and atrioventricular node. Projections of these postganglionic neurons terminate in the heart.
Brainstem and hypothalamus-derived supraspinal pathways control spinal sympathetic and parasympathetic outflow, thereby modulating hemodynamics. Previous research characterized five main brain regions providing input to SPNs in the thoracic spinal cord (Figure 1A; red fibers), including the rostral ventrolateral medulla (RVLM), the rostral ventromedial medulla, the caudal raphe nuclei, the A5 region of brainstem, and the paraventricular nucleus (PVN) of the hypothalamus. A phenotypically diverse group of neurons, e.g., adrenergic, glumatergic and GABAergic profiles, in the RVLM appear to play a major role in regulating cardiovascular function. They have been shown to tonically activate spinal sympathetic neurons and maintain arterial pressure and heart rate (Guyenet et al., 1989). Anatomical and physiological studies have demonstrated that neurons in the RVLM including the C1 group mainly give rise to supraspinal vasomotor pathways (Jeske and McKenna, 1992; Dampney et al., 2000). In addition, neurons in other supraspinal regions including the locus coeruleus, arcuate nucleus, and cortex also project to autonomic nuclei in the spinal cord. Experiments reported the discrepancies of descending vasomotor projections localization within the spinal cord among different species. Supraspinal vasomotor pathways are mainly located in the dorsal aspect of the lateral funiculus in humans (Nathan and Smith, 1987; Furlan et al., 2003), in extensive areas from ventral to dorsal aspects of the lateral funiculi in non-human primates, and between the dorsolateral sulcus and dental ligament in cats (Kerr and Alexander, 1964; Lebedev et al., 1986). In rodents, descending vasomotor projections have been identified within the dorsolateral aspects of the spinal cord by using electrophysiological techniques or neuronal tract tracing (Ruggiero et al., 1989; Strack et al., 1989). The supraspinal pathways innervating spinal SPNs contain a varied array of neurotransmitters including amino acids, catecholamines, and neuropeptides. The supraspinal cardiovascular regulation is further modulated by the limbic system.
Neurogenic Shock and Orthostatic Hypotension
The spinal cord enters a state referred to as spinal shock following an insult. During this period that can last from a few days up to 4-12 weeks in humans, reflexes and sensations are absent or depressed below the level of injury (Grigorean et al., 2009). Subsequently, neurogenic shock is experienced shortly after SCI at high levels. Individuals affected by neurogenic shock experience severe hypotension, bradycardia, and hypothermia as a result of the reduced sympathetic activity.
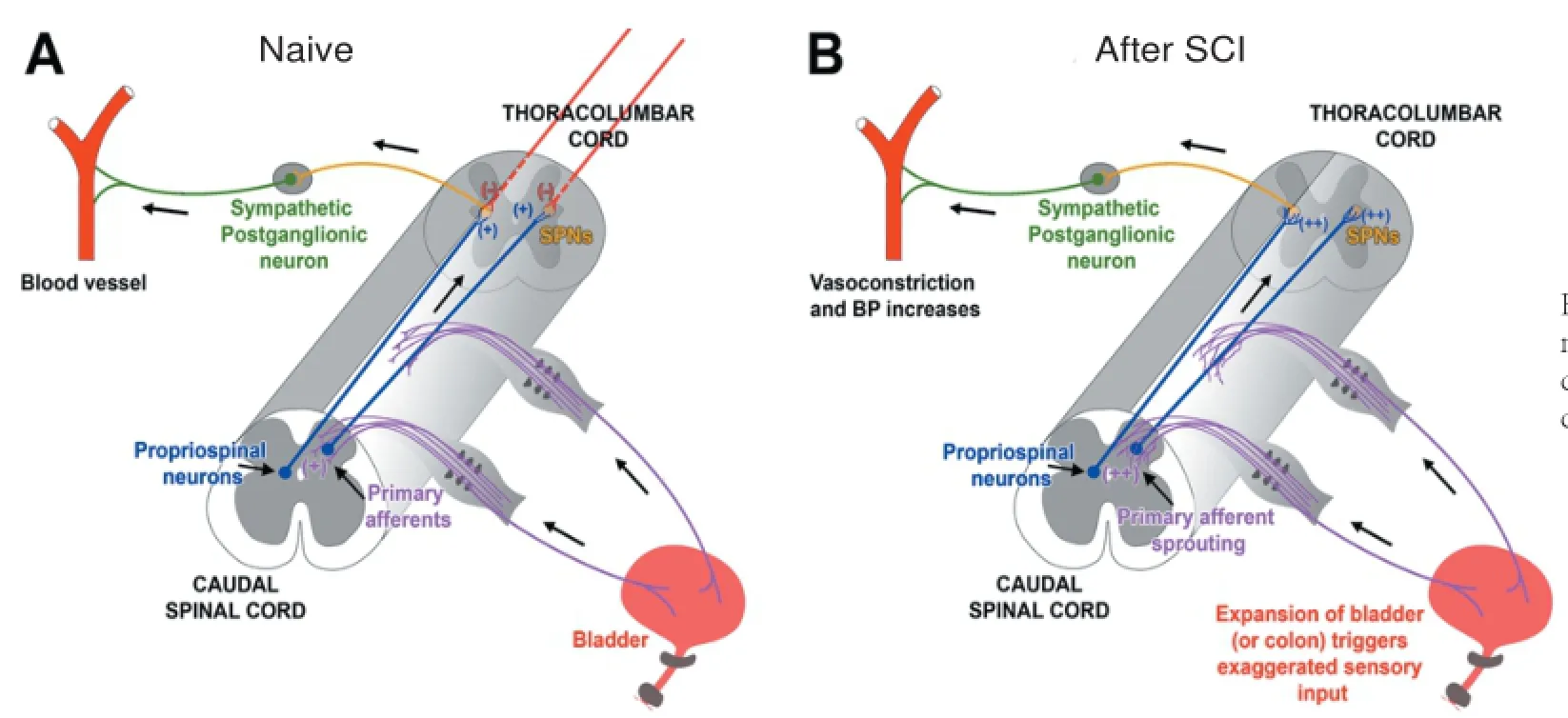
Figure 1 Schematic illustrating mechanisms involved in the development of autonomic dysreflexia (AD) following SCI.
Patients with acute or chronic SCI at a cervical or high thoracic level often have orthostatic hypotension, which is defined as an overall drop in baseline arterial blood pressure and bradycardia. These individuals may experience a decrease of at least 20 mmHg in baseline systolic blood pressure with an increase of at least 10 mmHg in baseline diastolic blood pressure upon a postural change, i.e., from a supine position to an upright position. In addition to a decrease in arterial blood pressure, some SCI patients may experience blurred vision,light-headedness, dizziness, fatigue, restlessness, and dyspnea. It has been suggested that factors involved in the occurrence of orthostatic hypotension include: (1) excessive venous pooling of blood in the organs and lower extremities due to reduced sympathetic activity and loss of reflexive vessel constriction below the injury level; (2) loss of lower extremity muscle function for counteracting venous pooling; (3) reduced plasma volume as a consequence of hyponatremia, an electrolyte disturbance in which the sodium ion concentration in the plasma is lower than normal; (4) cardiovascular deconditioning resulting from prolonged bed rest (Wecht et al., 2003). The degree of orthostatic hypotension in SCI patients may vary depending on the severity and level of the injury. For example, the extent of hypotension is greater in tetraplegics compared to paraplegics. The different timeframes of orthostatic hypotension following SCI may be due to the stages having distinct mechanisms. In the acute stage, the occurrence of orthostatic hypotension is mostly a result of losing supraspinal vasomotor tone and pooling of blood in the peripheral and splanchnic vasculature. However, orthostatic hypotension in the chronic stage is more related to the reduction of sympathetic activity below the level of injury. Also, the prolonged loss of muscle function below the level of injury contributes to blood pooling in the lower extremities to exacerbate hypotension.
Orthostatic hypotension can be treated physically and pharmacologically. The symptoms are possibly diminished under frequent postural change to a head-up position. Avoiding rapid postural changes can also be effective to prevent a drop in blood pressure. Because peripheral blood pooling is an important factor of the occurrence, in some cases, preventing postural-related venous dilation is useful to reduce hypotension by using abdominal binders, thigh cuffs and lower limb elastic stockings. Individuals with SCI usually do not need treatment with drugs for low blood pressure. If severe symptoms persist, though, a variety of drugs can be used including vasoconstrictors ephedrine, α-adrenergic receptor agonist midodrine, dopamine receptor blocker domperidone and others (Mathias, 2003). Clinicians must be careful with which treatments they choose because certain medications may exacerbate the effects of AD. It is of utmost importance that mechanisms behind AD should be understood when creating a treatment strategy for SCI patients.
Cardiac Dysrhythmias
In humans, the most common symptom in the acute stage after SCI is sinus bradycardia, though other cardiac dysrhythmias may occur, such as cardiac arrest, sinus tachycardia, supraventricular tachycardia, and atrial fibrillation. Almost all quadriplegic patients with a severe cervical SCI have persistent bradycardia, defined as mean heart rate for at least 1 day of <60 beats/min (Winslow et al., 1986). Though cardiac dysrhythmias emerge in acutely after severe SCI at high levels, cardiac performance can spontaneously improve over time. Atropine can partially and transiently treat bradycardia because of sympathetic deficiency. β-Adrenergic receptor agonists are used to eliminate the sinus pause (Lehmann et al., 1987). As a compensatory response to hypotension, following the period of neurogenic shock, heart rate usually increases in rat models of high-level SCI (Krassioukov and Weaver, 1995; Laird et al., 2006).
The Development of AD
AD is characterized by a sharp increase in blood pressure and baroreflex-mediated bradycardia in response to visceral or cutaneous stimuli below the level of SCI. In 1860, Hilton first described the condition of AD after observing the symptoms in a 21-year-old man with a complete SCI at vertebral level C5. Because of the SCI, the subject had bowel dysfunction, as his“bowels (did not) open without medicine. On some days he (had) peculiar sensation of chilliness, becoming pale, then (felt) hot and flushed both at defecation and micturition. The more constipated the bowel, the more these peculiar sensations (were) experienced.” Not much more was understood about AD until after World War I. In 1917, Riddoch and Head more extensively described AD symptoms in several case studies of men who acquired a SCI while in combat. These include excessive sweating, unchecked spinal reflexes, headaches, bradycardia, shivering, flushing, an increase in arterial pressure, piloerection, dysrhythmias and anxiety. Later, it was documented that cases of retinal, cerebral, or subarachnoid hemorrhages, and even death occurred after a rise in arterial blood pressure in individuals with SCI, indicating that AD can have very dire consequences. Indeed, patients who experience AD are at higher than normal risk for stroke, seizures, and other autonomic complications (Krassioukov and Claydon, 2006). While the majority of affected individuals with AD have high thoracic or cervical injuries (up to 90% of individuals with a lesion above T6), some patients with a spinal lesion as low as T10occasionally display symptoms similar to AD.
In patients with a high-level SCI, an annoying sensory stimulus, such as a blocked bladder catheter or pressure sore, may activate a sympathetic response, such as eliciting vasoconstriction of the splanchnic vascular bed below the level of injury and elevating blood pressure. Subsequently, increased blood pressure is sensed by baroreceptors in the carotid and aortic arch, which send projections up to the brainstem. To counteract the rise in blood pressure, the integration center in the brainstem transmits signals to the heart via parasympathetic pathways to reduce the heart rate. In able-bodied people or people with incomplete or low-level injuries, descending vasomotor pathways generate inhibitory influence on excited SPNs to decrease their activity. Due to the high level and severity of injury in the patients with SCI, however, sufficient descending modulatory projections no longer innervate the SPNs. Thus, the SPNs remain active and blood pressure stays elevated. Seemingly paradoxically, episodic hypertension and bradycardia occur simultaneously (Lindan et al., 1980; Karlsson, 1999). In addition, the excessive parasympathetic output above the level of injury causes vasodilation in the upper body and gives rise to headaches, flushing, sweating, and nasal congestion.
Clinical Issues Concerning SCI Patients with AD
A major clinical problem that many SCI patients experience is dysfunctional thermoregulation, including lower basal core temperatures and a compromised ability of the body located below the level of the injury to respond to changes in temperature (impaired ability to shiver when cold and to sweat when hot). The severity of the injury directly correlates with the de-gree of dysregulation and indirectly correlates with how much tissue is spared. Therefore, it is reasonable to use thermodysregulation to assess the extent of the injury as well as residual autonomic function.
Another issue, as earlier noted, is the loss of cardiovascular regulation. The inability to regulate BP and HR becomes particularly problematic when concerning physical performance of high-level athletes. Like most SCI patients, paralympic athletes typically have low BP that impairs their ability to pump blood and oxygen to taxed muscles during strenuous activities, such as competitive sports, compared to able-bodied people. Some paralympic athletes seek to enhance their performance in competition by increasing BP to compensate for the low BP. They often do so by self-inducing AD, also known as “boosting”, to take advantage of the strong sympathetic response (Mazzeo et al., 2015). Common methods of inducing AD include winding leg straps tightly (e.g., constricting feet, legs, scrotum, or testicles), bone fracture, or catheter locking to induce bladder swelling. While inducing repeated bouts of extreme hypertension may temporarily improve their athletic performance, it is at the detriment to their overall well-being, as discussed above. Thus, the International Paralympic Committee officially banned the practice.
Mechanisms behind the Development of AD
There are two phases of AD onset: as early as days or as late as weeks to months after injury that are likely a result of different mechanisms. AD that appears shortly after injury is likely a direct result of interrupting the descending projections to the SPNs below the injury. However, because fully developed AD may not manifest until at least 2 weeks after injury, it is clear that another mechanism contributes to the development of AD. The recording of renal sympathetic nerve activity suggests enhanced transmission through spinal reflex circuits after SCI (Maiorov et al., 1997). Furthermore, other studies using rodent animal models with complete SCI demonstrated that the reorganization of neuronal circuits in the injured spinal cord is a major contributor to the development of AD.
Intraspinal Plasticity Related to AD
One mechanism that is postulated to mediate AD is neuroplasticity that results in hyperexcitability of neural circuits below the level of injury. Below the lesion, the loss of descending neurotransmitter availability often triggers up-regulation of different receptors in spinal neurons, which augments the sensitivity. This process has been implicated in both spontaneous functional recovery and in the development of neurological symptoms, such as hyperreflexia (Miyazato et al., 2013). In support of this, rat spinal interneurons were found to be more excitable and sensitive to both innocuous and noxious cutaneous stimuli below a complete spinal transection site over time. Similarly, both innocuous and noxious somatic and visceral stimuli can elicit sympathetically-mediated AD. Moreover, greater increases in arterial pressure are triggered by colorectal distension (CRD) in chronically than acutely spinally-transected rats (Krassioukov et al., 2002).
The sprouting of pelvic primary afferents following SCI has been related to cardiovascular dysfunction (Figure 1A, B; purple fibers). The density of calcitonin gene-related peptide (CGRP)+afferent fibers increases at the lumbosacral spinal cord despite a lack of change in the number of counterpart DRG neurons (Krenz and Weaver, 1998b; Hou et al., 2009). This sprouting appears to be correlated with injury-induced elevation of nerve growth factor (NGF) in the spinal cord below the lesion although the mechanisms that underlie this plasticity are not completely understood. The level of NGF is much higher in the injured spinal cord than in the naïve. Because lumbosacral CGRP+fibers express the TrkA receptor, which binds to NGF, they are highly responsive to this growth factor. Administration of anti-NGF antibodies to prevent TrkA activation in the lower spinal cord after injury attenuates the fiber sprouting and diminishes AD (Krenz et al., 1999; Marsh et al., 2002). Likewise, if the chemorepellant semaphorin-3A is overexpressed in the dorsal horn of lumbar cord, both CGRP+fiber sprouting and AD are significantly reduced (Cameron et al., 2006). These data indicate that plasticity of the small-calibre nociceptive fibers plays a causative role in AD.
Reorganization of intraspinal circuits also likely contributes to alteration of input to sympathetic neurons. After SCI, denervated SPNs below the injury undergo short-term atrophy and lose dendritic arbors during the first week after injury. Nevertheless, normal SPN dendritic arbor and somal size can be re-established within 2 to 4 weeks after following the transient degenerative retraction (Krassioukov and Weaver, 1996; Krenz and Weaver, 1998a). Thus, it was proposed that synapses from supraspinal projections are replaced with new, interneuron inputs (Weaver et al., 1997). Neuroanatomical and electrophysiological evidence corroborate a significant role of propriospinal plasticity in the emergence of AD (Krassioukov et al., 2002). As pelvic primary afferents enter the lumbosacral spinal cord and cannot act directly on thoracolumbar SPNs, a propriospinal relay is believed to mediate the information of sprouted pelvic primary afferents to the thoracolumbar SPNs to elicit AD when the distal colon or bladder is distended (Figure 1A, B; blue fibers) (Rabchevsky, 2006). Using anterograde and retrograde neuronal tract tracing methods, both the density of lumbosacral propriospinal fibers and dorsal gray commissure (DGC) neurons significantly increase following complete high thoracic spinal cord transection. Furthermore, there is a significant increase in the number of c-Fos+cells in the lumbosacral gray matter of injured animals following noxious intermittent colorectal distension (Hou et al., 2008), indicating significant activation of local neural circuitry post to injury. Lastly, after spinal transection, spinal reflex circuits increase renal sympathetic nerve activities (Maiorov et al., 1997).
This intraspinal plasticity may be viewed as maladaptive and result in additional pathologies, such as neuropathic pain (either in the form of hyperalgesia or allodynia). Due to the severity of the injury, many AD patients do not always consciously feel the pain that is induced by nociceptive or non-nociceptive sensory stimuli. However, any sparing of spinothalamic or other sensory fibers that communicate pain could be problematic. Because of the sprouting, sensory circuitry after injury typically exhibits a greater degree of excitation than normal (Krassioukov et al., 2002). Primary afferent fibers that synapse in the superficial layers of the dorsal horn can exhibit robust increases in axon densities below the level of the lesion, potentially leading to the development of allodynia or hyperalgesia (Krenz et al., 1998b). This means that normally innocuous stimuli orsub-threshold noxious stimuli can trigger strong sympathetic responses and an AD episode.
Along these lines, though patients with severe, high-level SCI that are susceptible to AD are largely unable to perceive sensory stimuli delivered to their genitals, sensory circuits relaying input from the genitalia to the spinal cord remain intact. Thus, vibrational or electrical stimulation of the penis is able to trigger ejaculation and is often used to harvest sperm for fertility purposes. For reasons discussed above, such activation of hyperexcitable spinal sensory circuits triggers massive discharge of the SPN and often result in AD.
Changes to Peripheral Vasculature
Traumatic SCI affects the function of peripheral vasculature. The level of plasma catecholamines is often reduced in individuals with high SCI and blood vessels below the level of SCI become hypersensitive to α-adrenergic receptor (AR) stimulants (Krum et al., 1992). Infusing vasoconstrictors epinephrine and norepinephrine (NE) into individuals with a cervical SCI results in a higher increase in blood pressure than what is observed in non-injured individuals, suggesting that blood vessels become hypersensitive to NE (Mathias et al., 1976). In animal experiments, decentralization of postganglionic sympathetic neurons by cutting their preganglionic inputs results in a hypersensitive response to NE. In humans with quadriplegia and cervical SCI, enhanced pressor response to NE was also observed. Thus, peripheral α-AR hyper-responsiveness may partially account for a significantly enhanced pressor response during AD. This hyperreactivity to intrinsic and extrinsic factors are not the only change that occurs to blood vessels after SCI. Repeated bouts of hypertension following injury initiates a process of arterial remodeling. The blood vessels exhibit decreased arterial diameter and increased thickness of artery walls, making the arteries stiffer than the normal (Thijssen et al., 2012). This greatly affects blood flow and arterial pressure, and contributes to an increased risk of cardiovascular disease.
Potential Treatment Strategies
As previously described, SCI individuals undergo a myriad of acute complications, which in many cases, become chronic consequences of SCI. The treatment of SCI-induced cardiovascular disorders requires vigilance and active approaches to prevent or fully control its downstream effects. In the clinic, SCI individuals are usually subjected to constant monitoring of arterial blood pressure, heart rate, and any other signs that can aid clinicians determine to choose the appropriate treatment. Within 24-48 hours following SCI, admitted SCI patients are monitored for signs of severe hypotension and bradycardia that would indicate neurogenic shock (Hagen et al., 2011). In such cases, SCI individuals are treated accordingly with fluids, vasopressors (e.g., ephedrine), and atropine with adequate oxygenation if they exhibit a low heart rate. In addition to the implementation of pharmacological agents to control and maintain adequate mean arterial pressure levels, non-pharmacological strategies are also implemented to assist with orthostatic intolerance and help maintain arterial blood pressure at the appropriate level. Such strategies include avoiding or monitoring intake of substances that would contribute to orthostatic hypotension such as caffeine, alcohol, diuretics, salt and fluid intake, meal size, posture elevation, or any stressors that can result in vasodilation. SCI patients receive physical therapy and functional electrical stimulation if appropriate for their condition in an effort to further help control their arterial pressure, decrease venous pooling in the lower extremities and prevent orthostatic intolerance upon movement or a postural change.
To prevent the occurrence of AD, closely monitoring potential aversive stimuli that could elicit an episode is often exploited in the clinic. Such potential triggers include bowel or bladder distension and skin irritation (pressure sores, tight clothing, pressure on skin). Thus, one takes measures to preventative these triggers, such as draining the bladder. Once an AD episode begins develops, individuals are maintained in a position in which their head and trunk are elevated to avoid pooling of the blood. Pharmacological agents, e.g., vasodilators and anesthetics, are administered to prevent an increase in arterial blood pressure and assist in controlling or blocking an unfavorable sympathetic response. Because AD has been correlated with sprouting-associated hyperexcited circuits caudal to the injury, developing a strategy to counter this increased neural activity may treat the hypertensive response in SCI patients. Along these lines, administration of gabapentin, a pharmacological anticonvulsant that mimics the inhibitory neurotransmitter GABA that also affects calcium channel function, reduces spontaneous AD events, probably due to inhibition of the excitatory activity (Rabchevsky et al., 2011).
Another potential therapy is using cell transplantation to reestablish SCI-damaged neuronal pathways and restore function. Olfactory ensheathing cells were transplanted into the lesion of injured spinal cord at level T4(Kalincik et al., 2010). Although the researchers did not observe a difference in the degree of CRD-induced hypertension, there was a difference in the duration of the response - they found the hypertension attenuated faster in the experimental group than in the control group. Recently, rat embryonic brainstem- or spinal cord-derived neural stem cells were grafted into a complete spinal cord transection adult model (Hou et al., 2013). After eight weeks, animals grafted with brainstem-derived cells exhibited full recovery of basal hemodynamics, and both stem cell grafts contributed to mitigated AD in response to CRD. Histological analysis revealed graft-derived cells differentiated into catecholaminergic and serotonergic neurons that predominantly projected to SPNs in the IML below the injury, possibly establishing a compensatory relay. Additionally, because disrupted brainstem-derived descending axons appear more prone to regrow (given appropriate supporting substrates and stimulation) than descending tracts originating from cortex, we may eventually be able to use cell transplantation as a bridge to promote targeted, functional axon regeneration of brainstem populations that normally innervate SPN and reestablish descending control of sympathetic activity. Therefore, cell transplantation is a promising strategy to repair supraspinal vasomotor pathways and lead to cardiovascular functional recovery after SCI.
Conclusion
Attention to cardiovascular dysfunction after SCI is increasing. Though we know that main contribution factors to AD include the interruption of supraspinal vasomotor pathways, primary and propriospinal plasticity, and a hyperreflexic sympathetic response, a complete understanding of the pathophysiological mechanisms underlying AD is still lacking. Because of themulti-faceted etiology of AD, the most effective therapy to permanently restore cardiovascular function following SCI will likely be a combination approach that includes pharmacological agents, neural protection, axon regeneration, and removal of aberrant plasticity.
Author contributions: EP, EM, SH, and VJT wrote the paper; EM and VJT made the figure. All authors approved the final version of the paper.
Conflicts of interest: None declared.
Cameron AA, Smith GM, Randall DC, Brown DR, Rabchevsky AG (2006) Genetic manipulation of intraspinal plasticity after spinal cord injury alters the severity of autonomic dysreflexia. J Neurosci 26:2923-2932.
Dampney RA, Tagawa T, Horiuchi J, Potts PD, Fontes M, Polson JW (2000) What drives the tonic activity of presympathetic neurons in the rostral ventrolateral medulla? Clin Exp Pharmacol Physiol 27:1049-1053.
Furlan JC, Fehlings MG, Shannon P, Norenberg MD, Krassioukov AV (2003) Descending vasomotor pathways in humans: correlation between axonal preservation and cardiovascular dysfunction after spinal cord injury. J Neurotrauma 20:1351-1363.
Garshick E, Kelley A, Cohen SA, Garrison A, Tun CG, Gagnon D, Brown R (2005) A prospective assessment of mortality in chronic spinal cord injury. Spinal Cord 43:408-416.
Grigorean VT, Sandu AM, Popescu M, Iacobini MA, Stoian R, Neascu C, Strambu V, Popa F (2009) Cardiac dysfunctions following spinal cord injury. J Med Life 2:133-145.
Guyenet PG, Haselton JR, Sun MK (1989) Sympathoexcitatory neurons of the rostroventrolateral medulla and the origin of the sympathetic vasomotor tone. Prog Brain Res 81:105-116.
Hagen EM, Faerestrand S, Hoff JM, Rekand T, Gronning M (2011) Cardiovascular and urological dysfunction in spinal cord injury. Acta Neurol Scand Suppl 71-78.
Hou S, Duale H, Rabchevsky AG (2009) Intraspinal sprouting of unmyelinated pelvic afferents after complete spinal cord injury is correlated with autonomic dysreflexia induced by visceral pain. Neuroscience 159:369-379.
Hou S, Tom VJ, Graham L, Lu P, Blesch A (2013) Partial restoration of cardiovascular function by grafting embryonic neural stem cell grafts after complete spinal cord transection. J Neurosci 33:17138-17149.
Hou S, Duale H, Cameron AA, Abshire SM, Lyttle TS, Rabchevsky AG (2008) Plasticity of lumbosacral propriospinal neurons is associated with the development of autonomic dysreflexia after thoracic spinal cord transection. J Comp Neurol 509:382-399.
Jeske I, McKenna KE (1992) Quantitative analysis of bulbospinal projections from the rostral ventrolateral medulla: contribution of C1-adrenergic and nonadrenergic neurons. J Comp Neurol 324:1-13.
Kalincik T, Choi EA, Feron F, Bianco J, Sutharsan R, Hayward I, Mackay-Sim A, Carrive P, Waite PM (2010) Olfactory ensheathing cells reduce duration of autonomic dysreflexia in rats with high spinal cord injury. Auton Neurosci 154:20-29.
Karlsson AK (1999) Autonomic dysreflexia. Spinal Cord 37:383-391.
Kerr FW, Alexander S (1964) Descending autonomic pathways in the spinal cord. Arch Neurol 10:249-261.
Krassioukov A, Claydon VE (2006) The clinical problems in cardiovascular control following spinal cord injury: an overview. Prog Brain Res 152:223-229.
Krassioukov AV, Weaver LC (1995) Episodic hypertension due to autonomic dysreflexia in acute and chronic spinal cord-injured rats. Am J Physiol 268:2077-2083.
Krassioukov AV, Weaver LC (1996) Morphological changes in sympathetic preganglionic neurons after spinal cord injury in rats. Neuroscience 70:211-225.
Krassioukov AV, Johns DG, Schramm LP (2002) Sensitivity of sympathetically correlated spinal interneurons, renal sympathetic nerve activity, and arterial pressure to somatic and visceral stimuli after chronic spinal injury. J Neurotrauma 19:1521-1529.
Krenz NR, Weaver LC (1998a) Changes in the morphology of sympathetic preganglionic neurons parallel the development of autonomic dysreflexia after spinal cord injury in rats. Neurosci Lett 243:61-64.
Krenz NR, Weaver LC (1998b) Sprouting of primary afferent fibers after spinal cord transection in the rat. Neuroscience 85:443-458.
Krenz NR, Meakin SO, Krassioukov AV, Weaver LC (1999) Neutralizing intraspinal nerve growth factor blocks autonomic dysreflexia caused by spinal cord injury. J Neurosci 19:7405-7414.
Krum H, Howes LG, Brown DJ, Ungar G, Moore P, McNeil JJ, Louis WJ (1992) Risk factors for cardiovascular disease in chronic spinal cord injury patients. Paraplegia 30:381-388.
Laird AS, Carrive P, Waite PM (2006) Cardiovascular and temperature changes in spinal cord injured rats at rest and during autonomic dysreflexia. J Physiol 577:539-548.
Lavdas AA, Blue ME, Lincoln J, Parnavelas JG (1997) Serotonin promotes the differentiation of glutamate neurons in organotypic slice cultures of the developing cerebral cortex. J Neurosci 17:7872-7880.
Lebedev VP, Krasyukov AV, Nikitin SA (1986) Electrophysiological study of sympathoexcitatory structures of the bulbar ventrolateral surface as related to vasomotor regulation. Neuroscience 17:189-203.
Lee BB, Cripps RA, Fitzharris M, Wing PC (2014) The global map for traumatic spinal cord injury epidemiology: update 2011, global incidence rate. Spinal cord 52:110-116.
Lehmann KG, Lane JG, Piepmeier JM, Batsford WP (1987) Cardiovascular abnormalities accompanying acute spinal cord injury in humans: incidence, time course and severity. J Am Coll Cardiol 10:46-52.
Lindan R, Joiner E, Freehafer AA, Hazel C (1980) Incidence and clinical features of autonomic dysreflexia in patients with spinal cord injury. Paraplegia 18:285-292.
Loewy AD, Spyer KM (1990) Central regulation of autonomic functions. New York: Oxford University Press.
Maiorov DN, Weaver LC, Krassioukov AV (1997) Relationship between sympathetic activity and arterial pressure in conscious spinal rats. Am J Physiol 272:H625-631.
Marsh DR, Wong ST, Meakin SO, MacDonald JI, Hamilton EF, Weaver LC (2002) Neutralizing intraspinal nerve growth factor with a trkA-IgG fusion protein blocks the development of autonomic dysreflexia in a clip-compression model of spinal cord injury. J Neurotrauma 19:1531-1541.
Mathias CJ (2003) Autonomic diseases: management. J Neurol Neurosurg Psychiatry 74 Suppl 3:iii42-47.
Mathias CJ, Frankel HL, Christensen NJ, Spalding JM (1976) Enhanced pressor response to noradrenaline in patients with cervical spinal cord transection. Brain 99:757-770.
Mazzeo F, Santamaria S, Iavarone A (2015) “Boosting” in paralympic athletes with spinal cord injury: doping without drugs. Funct Neurol 30:91-98. Miyazato M, Oshiro T, Chancellor MB, de Groat WC, Yoshimura N, Saito S (2013) An alpha1-adrenoceptor blocker terazosin improves urine storage function in the spinal cord in spinal cord injured rats. Life Sci 92:125-130.
Nathan PW, Smith MC (1987) The location of descending fibres to sympathetic preganglionic vasomotor and sudomotor neurons in man. J Neurol Neurosurg Psychiatry 50:1253-1262.
Rabchevsky AG (2006) Segmental organization of spinal reflexes mediating autonomic dysreflexia after spinal cord injury. Prog Brain Res 152:265-274.
Rabchevsky AG, Patel SP, Duale H, Lyttle TS, O’Dell CR, Kitzman PH (2011) Gabapentin for spasticity and autonomic dysreflexia after severe spinal cord injury. Spinal Cord 49:99-105.
Ruggiero DA, Cravo SL, Arango V, Reis DJ (1989) Central control of the circulation by the rostral ventrolateral reticular nucleus: anatomical substrates. Prog Brain Res 81:49-79.
Strack AM, Sawyer WB, Hughes JH, Platt KB, Loewy AD (1989) A general pattern of CNS innervation of the sympathetic outflow demonstrated by transneuronal pseudorabies viral infections. Brain Res 491:156-162.
Thijssen DH, De Groot PC, van den Bogerd A, Veltmeijer M, Cable NT, Green DJ, Hopman MT (2012) Time course of arterial remodelling in diameter and wall thickness above and below the lesion after a spinal cord injury. Eur J Appl Physiol 112:4103-4109.
Weaver LC, Cassam AK, Krassioukov AV, Llewellyn-Smith IJ (1997) Changes in immunoreactivity for growth associated protein-43 suggest reorganization of synapses on spinal sympathetic neurons after cord transection. Neuroscience 81:535-551.
Wecht JM, De Meersman RE, Weir JP, Spungen AM, Bauman WA (2003) Cardiac autonomic responses to progressive head-up tilt in individuals with paraplegia. Clin Auton Res 13:433-438.
Winslow EB, Lesch M, Talano JV, Meyer PR, Jr. (1986) Spinal cord injuries associated with cardiopulmonary complications. Spine (Phila Pa 1976) 11:809-812.
*Correspondence to: Veronica J. Tom, Ph.D., veronica.tom@drexelmed.edu.
orcid: 0000-0002-3369-7575 (Veronica J. Tom)
10.4103/1673-5374.177707 http://www.nrronline.org/
Accepted: 2016-01-23
How to cite this article: Partida E, Mironets E, Hou S, Tom VJ (2016) Cardiovascular dysfunction following spinal cord injury. Neural Regen Res 11(2):189-194.
Funding: This work was supported by research grants to VJT from the National Institutes of Health (R01 NS085426) and the Department of Defense (W81XWH-14-1-060).
杂志排行

中国神经再生研究(英文版)的其它文章
- Gas6-Tyro3 signaling is required for Schwann cell myelination and possible remyelination
- Impaired consciousness caused by injury of the lower ascending reticular activating system: evaluation by diffusion tensor tractography
- Tissue-type plasminogen activator is a modulator of the synaptic vesicle cycle
- Practical application of the neuroregenerative properties of ketamine: real world treatment experience
- Exergames: neuroplastic hypothesis about cognitive improvement and biological effects on physical function of institutionalized older persons
- Neurotrophic factor therapy for Parkinson’s disease: past, present and future