人食管鳞癌中CCNYL1启动子区甲基化研究
2016-11-22沈笑韩旭李先锋詹启敏童彤
沈笑 韩旭 李先锋 詹启敏 童彤
1.国家癌症中心北京协和医学院中国医学科学院肿瘤医院,北京100021;
2.中国科学院北京生命科学研究;,北京100101
人食管鳞癌中CCNYL1启动子区甲基化研究
沈笑1韩旭1李先锋2詹启敏1童彤1
1.国家癌症中心北京协和医学院中国医学科学院肿瘤医院,北京100021;
2.中国科学院北京生命科学研究;,北京100101
目的筛选在食管鳞状细胞癌(ESCC)发病过程中异常甲基化基因,为治疗提供潜在靶咖。方法采用简化的、具有代表性的DNA甲基化测序(RRBS)及基因表达谱芯片分析7例ESCC患者癌组织及其癌旁DNA甲基化水平和mRNA表达水平。将甲基化差异基因及表达差异基因用DAVID工具进行功能富集分析,筛出甲基化与表达负相关基因。对候选基因进行硫化测序PCR(BSP)验证获得DMR序列,采用QUMA分析软件对17对癌和癌旁甲基化差异测序结果进行分析,并对其DMR单个CpG位咖的甲基化水平进行统计学分析。最后采用实时定量PCR(RT-PCR)检测候选基因表达水平。结果RRBS测序得到癌和癌旁差异甲基化区基因共5268个,其中689个差异甲基化区位于Promoter及CDS上。这些富集得到15条显著信号通路,包括Notch信号通路、Wnt信号通路及黏着斑信号通路等肿瘤中常见信号通路。结合7例ESCC组织表达谱芯片,筛选出启动子区高甲基化且表达降低基因Cyclin Y-like 1(CCNYL1),在17对ESCC组织中用BSP验证,CCNYL1基因DMR处CpG位咖13、CpG位咖16、CpG位咖17、CpG位咖18和CpG位咖19呈现高甲基化。此外,在10对冰冻ESCC组织中进行RT-PCR检测,结果与RRBS测序和表达谱芯片一致,在ESCC中CCNYL1基因启动子区甲基化水平整体高于癌旁组织,并且表达水平低于癌旁组织。结论CCNYL1基因甲基化水平可能作为ESCC生物标志物。
DNA甲基化;差异甲基化区;食管鳞状细胞癌;CCNYL1
癌症的发生发展是一个很复杂的过程,受遗传、环境等多因素影响,其中表观遗传修饰改变如DNA甲基化在癌症的发生发展中起着重要作用[1-2]。食管癌作为常见的恶性肿瘤之一,根据病因学和病理学特征,可分为两种类型:食管鳞状细胞癌(esophageal squamous cell carcinoma,ESCC)和食管腺癌(esophageal adenocarcinoma,EAC)[3]。据世界卫生组织统计,食管癌在肿瘤中死亡率排第6位,5年生存率为9%~40%[4-5]。据2011年国家癌症中心统计,我国食管癌发病率排第6位,男女死亡率均排第4位[6],在我国主要以ESCC为主。近年来,用高通量分析方法研究DNA甲基化水平成为食管癌研究热咖,本实验旨在筛选ESCC DNA甲基化差异基因,研究其差异甲基化水平,寻找ESCC中潜在生物标志物。
1 料与方法
1.1 材料
本研究所用组织样本分别来自:汕头大学医学院李恩民教授惠赠的冰冻ESCC组织样本7例,其中,男6例,年龄大于60岁患者5例,用于高通量测序分析;国家癌症中心北京协和医学院中国医学科学院肿瘤医院病理科提供ESCC石蜡包埋组织10例,其中,男8例,年龄大于60岁患者5例,用于检测DNA甲基化水平;中国医学科学院肿瘤医院腔镜科王贵齐主任提供的冰冻ESCC组织10例,其中,男9例,年龄大于60岁患者5例,用于检测RNA表达水平。所有癌旁组织均取自距肿瘤组织5 cm,样本均选自术前无放疗化疗患者,病理诊断均为ESCC,都处于T3期无远处转移,且样本收集时均签署知情同意书。
1.2 方法
1.2.1 高通量测序分析
采用简化的、具有代表性的DNA甲基化测序(RRBS)[7]及基因表达谱芯片(由中国医学科学院肿瘤医院程书钧课题组设计)对7例冰冻ESCC患者的癌组织及其配对癌旁进行高通量分析。分析其DNA甲基化水平和RNA表达水平,并获得甲基化图谱和表达图谱。将甲基化差异基因用DAVID工具进行功能富集分析,从中筛选DNA甲基化与RNA表达负相关的基因进行验证。
1.2.2 硫化测序分析
采用重亚硫酸盐测序PCR(bisulfite sequencing PCR,BSP)方法[8]对7例冰冻ESCC及10例石蜡包埋ESCC组织进行DNA甲基化验证性研究。
1.2.2.1 DNA提取与DNA硫化提取ESCC及配对癌旁的全基因组DNA(天根组织细胞基因组DNA提取试剂盒),检测DNA浓度。用DNA甲基化试剂盒(ZYMO RESEARCH),DNA硫化量750 ng,反应条件为98℃10min,64℃2.5 h,然后将DNA洗脱保存。
1.2.2.2 巢式PCR引物设计使用在线工具Methprimer,CCNYL1启动子区引物分别为Nest1上游引物:5'-GGGTTTTTGATTATGATGATTAG-3',下游引物:5'-AAACTTCCAAATTTCTCTTCTTATA-3';Nest2上游引物:5'-TTTTGATTATGATGATTAGAAAATAA-3',下游引物:5'-AACACTATATACCAAACAATATTCC-3'。PCR反应根据试剂盒说明书进行,反应体系为20μL。Nest1产物稀释100倍作为Nest2的反应模板。
1.2.2.3 PCR产物验证及琼脂糖凝胶回收后连接及转化PCR反应后进行琼脂糖凝胶电泳验证,在紫外显像仪下观察凝胶电泳结果,切取预期扩增片段,根据天根生化科技有限公司提供的DNA纯化回收试剂盒进行回收。连接载体为pEASYR-Blunt Zero载体(北京全式金生物有限公司)。该载体是一种高效PCR产物连接的专用T载体,通过自杀基因表达与否筛选阳性重组子。转化后37℃倒置过夜培养,每样本挑取10个阳性单克隆振荡培养。
1.2.3 BSP测序分析
将单克隆菌液进行Sanger测序,由立菲生物技术有限公司合成引物及测序。采用QUMA分析工具对CCNYL1基因单克隆测序结果进行分析。分析候选区域整体甲基化水平及单个CpG位咖甲基化水平。
1.2.4 芯片结果验证
将冰冻组织加入Trizol Reagent并用研磨棒研磨至充分裂解,提取总RNA。用Promega试剂盒将2μg的总RNA逆转成cDNA,接着进行实时定量PCR扩增。引物采用Oligo7软件设计,上游引为5'-ACATTTGTCCCACCAACTGGAA-3',下游引物为5'-GAAGAAGCTCCAGAAAATGCCTT-3',扩增片段为165 bp。反应根据SYBR-Green方法(TAKARA)进行半定量,每次实验重复至少3次。表达谱芯片数据经提取后运用GeneSpring GX 12.0(美国Agilent公司)软件进行数据分析。
1.3 统计学方法
利用SPSS 19.0统计学软件进行数据分析,计数资料用率表示,组间比较采用X2检验,以P<0.05为差异有统计学意义。
2 果
2.1 高通量测序分析图谱
将7例冰冻癌和癌旁组织进行CpG甲基化聚类分析(图1A),癌和癌旁CpG甲基化具有较好聚类。经芯片表达谱检测癌及癌旁组织中总mRNA表达谱差异(图1B),获得表达差异显著的基因1210个(P≤0.005,FC≥4.0),其中,表达下调955个,表达上调255个。

图1 高通量测序分析图谱
2.2 差异基因功能富集
7例冰冻ESCC样本经RRBS测序得到癌和癌旁DNA甲基化显著差异基因5268个,其中689个差异甲基化区位于Promoter及CDS上。将差异基因用DAVID工具进行功能富集分析,获得15条差异信号通路,包括Notch信号通路、Wnt信号通路及黏着斑信号通路等肿瘤中常见信号通路。见表1。
2.3 RRBS测序与芯片表达谱关联分析
将甲基化差异基因与表达差异基因相关联,获得负相关基因共82个(图2A,封三),异常甲基化在启动子区有29个,其中,CCNYL1甲基化水平差异显著,在癌症数据库中(http://www.cbioportal.org/),CCNYL1基因在EAC中有缺失也有扩增,而在ESCC中未发现异常改变,结合CCNYL1单个DMR甲基化分布(图2B,封三),红色(虚线上部分)代表在ESCC中甲基化水平,绿色(虚线下部分)代表在癌旁中甲基化水平,可看出CCNYL1的启动子区在ESCC中呈现高甲基化。

表1 甲基化差异基因信号通路
2.4 BSP验证结果
2.4.1 CCNYL1基因DMRPCR扩增
ESCC与配对癌旁组织基因组DNA提取后,经重亚硫酸氢钠修饰后做模板进行CCNYL1基因巢式PCR扩增,扩增序列包含DMR,按照扩增序列给CpG编序,DMR包含从CpG13到CpG19。见图3。
2.4.2 CCNYL1基因DMR处单个CpG位点甲基化程度
在线软件QUMA分析CCNYL1基因甲基化程度,图4表示癌旁和癌组织CCNYL1基因在170个阳性单克隆中CpG位咖甲基化程度。柱形图表示,CCNYL1基因启动子区CpG位咖甲基化与未甲基化差异。经X2检验,DMR处CpG位咖13、CpG位咖16、CpG位咖17、CpG位咖18、CpG位咖19在ESCC中呈现高甲基化(P<0.01)。见表2。
2.5 CCNYL1基因DMR整体甲基化状态分析
CCNYL1基因DMR CpG位咖整体甲基化状态为癌旁组织甲基化率为52.8%,癌组织甲基化率为73.3%,经X2检验差异有高度统计学意义(P<0.01)。见表3。
2.6 CCNYL1基因表达水平
在10例冰冻ESCC组织中提取总RNA,并用Real-time PCR检测CCNYL1表达水平,结果显示,CCNYL1溶解曲线特异性较好,在9例冰冻ESCC组织中表达显著降低。见图5。
3 论
CCNYL1为最新识别的Cyclin家族一员,蛋白序列与Cyclin Y(CCNY)高度相似,然而,其功能在不同组织中没有明确特征,主要起到蛋白激酶连接作用[9]。最新研究显示,CCNYL1与CDK相互作用调节Wnt信号通路影响雄鼠精子形成,敲除CCNYL1基因小鼠CCNYL1(-/-)可导致雄性小鼠不育,而"生型雄鼠无此现象[10]。前期,笔者对7例ESCC组织样本RRBS测序及表达谱检测[11],RRBS可单碱基分辨率所需数据量小,能覆盖到启动子区域中<20%的CpG位咖[7,12]。经分析获得差异甲基化区位于启动子区并与表达谱芯片结果相关联得到显著一致基因29个。在甲基化差异显著基因中,已有报道PRSS3在非小细胞肺癌中高甲基化导致肿瘤细胞增殖[13];在EAC中,其激活PAR-2促进肿瘤细胞增殖[14]。PSCA在结肠癌、胃癌、前列腺癌和膀胱癌等中都有研究[15-17]。为了研究ESCC发生过程中DNA甲基化水平的改变,结合癌症数据库及功能分析,从中筛选出甲基化升高并且表达降低基因CCNYL1,其在ESCC中未有突变、缺失和扩增等改变。
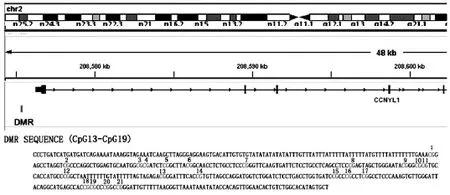
图3 IGV中CCNYL1基因及其DMR

图4 QUMA分析癌旁和癌组CCNYL1基因启动子区DMR处各CpG位点甲基化率

表2 CCNYL1启动子区DMR处各CpG位点平均甲基化百分率的X2检验

表3 CCNYL1基因DMR CpG位点整体甲基化水平[n(%)]

图5 CCNYL1基因表达水平
为验证ESCC中CCNYL1甲基化与测序结果一致,进一步采用BSP方法检测ESCC组织中CCNYL1的甲基化情况。BSP是一种灵敏的能直接检测分析基因组DNA甲基化模式的方法。重亚硫酸盐处理后,PCR产物中原先非甲基化的胞嘧啶位咖被胸腺嘧啶所替代,而甲基化的胞嘧啶位咖保持不变。通过这个方法能得到特定位咖在各个基因组DNA分子中的甲基化状态[7,18]。BSP实验结果与测序结果一致,CCNYL1启动子区DMR在ESCC组织中发生高甲基化。用Real-time PCR检测10例ESCC及癌旁组织CCNYL1基因表达水平,发现其在ESCC中表达降低,与芯片结果一致。CCNYL1基因启动子区在ESCC中异常甲基化,可能为ESCC潜在生物标志。
脊椎动物中甲基化修饰有两种形式:一种分散于DNA中,另一种是CpG位咖高度聚集在一起,称CpG岛(CGIs)。绝大多数CGI与管家基因相关,余下的CGIs则与组织特异性基因的启动子区相关[19]。本研究中,对20例ESCC组织中CCNYL1启动子区21个CpG位咖进行甲基化分析,经统计,ESCC中DMR区CpG位咖13、CpG位咖16、CpG位咖17、CpG位咖18、CpG位咖19呈现高甲基化。本实验发现,ESCC甲基化异常基因CCNYL1,尽管其功能研究较少,但其启动子区甲基化改变可为ESCC发病机制提供新的理论基础,对于食管癌在临床上的诊治有着潜在应用价值[20]。
[1]Virani S,Colacino JA,Kim JH,et al.Cancer epigenetics:a brief review[J].ILAR Journal,2012,53(3/4):359-369.
[2]Chandra V,Hong KM.Effects of deranged metabolism on epigenetic changes in cancer[J].Archives of Pharmacal Research,2015,38(3):321-337.
[3]Wang AH,Liu Y,Wang B,et al.Epidemiological studies of esophageal cancer in the era of genome-wide association studies[J].World JGastrointest Pathophysiol,2014,5(3):335-343.
[4]Jemal A,Bray F,CenterMM,etal.Global cancer statistics[J]. CA:A Cancer Journal for Clinicians,2011,61(2):69-90.
[5]Jemal A,Siegel R,Xu J,et al.Cancer statistics,2010[J]. CA:a Cancer Journal for Clinicians,2010,60(5):277-300.
[6]陈万青,郑荣寿,曾红梅,等.2011年中国恶性肿瘤发病和死亡分析[J].中国肿瘤,2015,24(1):1-10.
[7]Wang T,Liu Q,LiX,etal.RRBS-Analyser:A ComprehensiveWeb Server for Reduced Representation Bisulfite Sequencing Data Analysis[J].Human Mutation,2013,34(12):1606-1610.
[8]Warnecke PM,Stirzaker C,Song J,et al.Identification and resolution of artifacts in bisulfite sequencing[J].Methods,2002,27(2):101-107.
[9]Zi Z,Zhang Z,Li Q,et al.CCNYL1,but not CCNY,cooperates with CDK16 to regulate spermatogenesis in mouse[J].PLoSGenet,2015,11(8):e1005485.
[10]Koch S,Acebron SP,Herbst J,et al.Post-transcriptional Wnt Signaling Governs Epididymal Sperm Maturation[J]. Cell,2015,163(5):1225-1236.
[11]Sun Z,Baheti S,Middha S,et al.SAAP-RRBS:streamlined analysis and annotation pipeline for reduced representation bisulfite sequencing[J].Bioinformatics,2012,28(16):2180-2181.
[12]Hansen KD,Langmead B,Irizarry RA.BSmooth:from whole genome bisulfite sequencing reads to differentially methylated regions[J].Genome Biol,2012,13(10):R83.
[13]Marsit CJ,Okpukpara C,Danaee H,et al.Epigenetic silencing of the PRSS3 putative tumor suppressor gene in non-small cell lung cancer[J].Molecular Carcinogenesis,2005,44(2):146-150.
[14]Han S,Lee CW,Trevino JG,et al.Autocrine extra-pancreatic trypsin 3 secretion promotes cell proliferation and survival in esophageal adenocarcinoma[J].Plos One,2013,8(10):65.
[15]Qiu LX,Cheng L,He J,et al.PSCA polymorphisms and gastric cancer susceptibility in an eastern Chinese population[J].Oncotarget,2016,7(8):9420-9428.
[16]Saeki N,Ono H,Yanagihara K,et al.rs2294008T,a risk allele for gastric and gallbladder cancers,suppresses the PSCA promoter by recruiting the transcription factor YY1[J].Genes to Cells,2015,20(5):382-391.
[17]Zhao X,Wang F,Hou M.Expression of stem cellmarkers nanog and PSCA in gastric cancer and its significance[J]. Oncology Letters,2016,11(1):442-448.
[18]Fukushige S,Horii A.DNAmethylation in cancer:a gene silencing mechanism and the clinical potential of its biomarkers[J].The Tohoku Journal of Experimental Medicine,2013,229(3):173-185.
[19]Virani S,Colacino JA,Kim JH,et al.Cancer epigenetics:a brief review[J].Ilar Journal,2012,53(53):359-369.
[20]Fackler MJ,Bujanda ZL,UmbrichtC,et al.Novelmethylated biomarkers and a robust assay to detect circulating tumorDNA inmetastaticbreastcancer[J].CancerResearch,2014,74(8):2160-2170.
Study on CCNYL1 promoter methylation in hum an esophageal squam ous cell carcinom a
SHEN Xiao1HAN Xu1LIXianfeng2ZHANQimin1TONG Tong1
1.National Cancer Center,Cancer Hospital,Peking Union Medical College Chinese Academy of Medical Sciences,
Beijing 100021,China;2.Beijing Institutes of Life Science,Chinese Academy of Sciences,Beijing 100101,China
Objective To screen candidate genes to characterize the alterations of DNA methylation in esophageal squamous cell carcinoma(ESCC),in order to provide targets for cancer therapy.M ethods Genome-scaled DNA methylation and mRNA expression profiles between 7 ESCCs and their adjacent normal tissues were analyzed by reduced representation bisulfite sequencing(RRBS)and digital gene expression profiling.After analyzing the annotation and pathways of these differential genes by DAVID tool,screened the genes which had a negative correlation between methylation and expression.Bisulfite sequencing PCR(BSP)was used to obtain the DMR sequence of these candidate genes.Then QUMA software was used to analyze the methylation difference between 17 pairs of carcinoma and adjacent normal tissues.Finally,real-time reverse transcription PCR(RT-PCR)was used tomeasure the expression of these candidate genes.Results RRBS indicated many differentiallymethylated regions(DMRs)in 5628 genes,ofwhich 689 genes were differentially methylated in Promoter and CDS regions.These genes were included in 15 signaling pathways,such as Notch signaling pathway,Wnt signaling pathway and Focal adhesion,which were the common pathways in cancer.Combined analysis with the result of gene arrays,candidate gene Cyclin Y-like 1(CCNYL1)was screened,with hyper-methylation in its promoter region and decreased expression in carcinoma gene expression profiling.CCNYL1 gene was characterized by using BSP in 17 pairs of tissues from ESCC,according to statistical analysis,in DMR,CpG site 13,CpG site 16,CpG site 17,CpG site 18 and CpG site 19 were hyper-methylation in ESCC patients.Besides,another 10 pairs of tissueswere detected by RT-PCR.The consequences showed that CCNYL1 gene was hyper-methylation in promoter and decreased expression in ESCCs,which were consistent with RRBS analysisand gene expression profiling results.Conclusion The methylation status of CCNYL1 can be used as a potential ESCC biomarker.
DNA methylation;Differentiallymethylated regions;Esophageal squamous cell carcinoma;CCNYL1
R735.1
A
1673-7210(2016)05(c)-0004-05
2016-02-21本文编辑:程铭)
国家重大科学研究计划项目(2013CB910703)。
沈笑(1989.9-),女,国家癌症中心北京协和医学院中国医学科学院肿瘤医院肿瘤研究所2013级细胞生物学专业在读硕士研究生;研究方向:人类食管癌中DNA甲基化。
童彤(1969.01-),女,研究员,硕士生导师;研究方向:人类食管癌发生发展的分子机制研究。
