生物大数据中的聚类方法分析
2016-09-21路东方许俊富项超娟上海大学计算机工程与科学学院上海200444
路东方,许俊富,项超娟,谢 江(上海大学计算机工程与科学学院,上海200444)
生物大数据中的聚类方法分析
路东方,许俊富,项超娟,谢江
(上海大学计算机工程与科学学院,上海200444)
随着人类基因组计划的实施和完成,生物实验技术快速发展,生物数据呈现爆发式增长并不断积累,生命科学迎来了大数据时代.在后基因组时代,单一的统计模式逐渐被智能化与综合分析相结合的方式所取代,聚类分析便是核心的数据挖掘方式.描述了生物信息学领域中的大数据现状,总结基因表达谱分析和生物网络分析中常用的聚类方法,并对小鼠胚胎成纤维细胞的时间序列数据进行实验对比.实验结果表明,不同的聚类方法生成了不同的实验结果,面临高噪声的生物大数据,选择或结合合适的聚类方法进行综合分析将有助于获得更可靠的分析结果.
生物大数据;数据分析;聚类方法
1 概述
生物信息学是一门交叉学科,包含了对生物信息数据的获取、处理、储存、转发、分析和解释等方方面面.生物信息学综合运用数学、计算机科学和生物学的各种工具,来阐明和理解生物信息数据所包含的生物学意义[1].生物信息学发展呈现出两个主要特征:一是伴随着海量生物数据的产生;二是相关科研活动逐步从传统的手工统计方式转向智能化与综合分析相结合的方式.
2000年6月26日,被誉为生命“阿波罗计划”的人类基因组计划工作草图的完成,是生物信息学发展史上又一个里程碑式的事件,它预示着完成人类基因组计划已经指日可待.迄今已完成了约40多种生物的全基因组测序工作,其中人类基因组已完成了约3×109个碱基对的测序任务.尤其是第二代测序技术[2]的发展,使得基因组学每天都产生数以“T”计的海量数据.
在数据爆炸时代,根据数据类型进行有效整合是非常重要的.截至2012年[3],仅记录在美国GenBank数据库(美国国家生物技术信息中心的DNA序列总数据库)中的DNA序列总量就已超过了70亿个碱基对[3-4].在这样一个生物信息的浪潮之巅,生物大数据的积累无疑会随着时间的演进而愈发增多.基于cDNA序列测序建立起来的reptiledatabase[5](爬行动物数据库)中已经存储了超过1万个基因的数据.美国国家生物技术信息中心(National Center for Biotechnology Information,NCBI)下的基因表达数据库(Gene Expression Omnibus,GEO)[6]中已存储了3 848个数据集,包括1 618 438个样本的基因表达、基因芯片、蛋白质结构信息等数据.在这些数据的基础上派生、整理出来的数据库已超过500个,这一切构成了生物学数据的海洋.这些科学数据的急速增长和海量积累[7],在人类的科学研究历史上是空前的.图1统计了GEO中从2000年到2015年存储样本数据的变化情况.

图1 GEO样本数据增长趋势Fig.1 Trends of GEO samples
常用的生物数据库分为以下三类:一是基于DNA序列的数据库,包括与DNA的复制、转录、修复等密切相关的蛋白质因子数据库,有GenBank、EMBL(European Molecular Biology Laboratory,欧洲分子生物学实验室的DNA和RNA序列库)、RepBase(真核生物DNA中重复序列数据库)等;二是基于核酸序列的数据库,主要有INSD(International Nucleotide Sequence Database,国际核酸序列数据库)、DDBJ(DNA Data Bank of Japan,日本核酸数据库)等;三是基于蛋白质建立的数据库,主要有BIOGRID(Biological General Repository for Interaction Datasets,蛋白质-蛋白质以及基因相互作用的数据库)、DIP(Database of Interacting Proteins,蛋白质功能和蛋白质-蛋白质关系数据库),STRING(Search Tool for the Retrieval of Interacting Genes/Proteins,已知和预测蛋白质相互作用数据库)、Uniprot(Universal Protein Resource,蛋白质序列功能信息数据库)等.
在当前的大数据时代,通常这些数据表现出以下四个特征[8-11]:①数据量巨大;②数据类别多样;③数据潜在价值高;④增长速度惊人.人类基因组计划的实施和完成,以及下一代测试技术(又称为高通量测序技术)的不断发展,不仅解决了生命科学发展中的经济障碍,而且产生了更多的生物数据,推进了生命科学乃至医学水平的发展.常见的生物数据类型包括基因组学、转录组学、蛋白质组学、代谢组学等,具体表现形式有基因的表达值、蛋白质相互作用、蛋白质结构等.生命科学的巨量数据正以多种类型和不同表现形式急剧膨胀,生命科学迎来了大数据时代[12-13].毫无疑问,这些生物大数据蕴涵着巨大价值,然而数据不等于信息和知识,而只是信息和知识的源泉.生物学家们需要将看似杂乱的数据转变成可用的资源以后才能充分利用这些数据.计算科学的发展在伴随着一定挑战的同时,也给生命科学的“大数据时代”带来了曙光.一方面,通过足够的技术保障,可以保存这些具有巨大价值的生物大数据;另一方面,通过一些特定的方法充分挖掘这些大数据中隐藏的信息,将给生命科学研究带来无限可能.
生物数据挖掘是当前生物数据研究的重点内容之一.常用的统计学方法已无法适应当前的大数据时代,而作为机器学习领域的无监督算法——聚类,却备受青睐.在生物信息学研究领域中,从基本的序列分析、分子进化和比较基因组学,到蛋白质结构比对和预测,再到计算机辅助药物设计等,聚类分析都显示出极大的优势.合适的聚类方法有助于发现相似的DNA,RNA或蛋白质功能组.一方面,是基于基因表达谱的聚类,其主要研究方式分为3种[14-15]:一是基于时间序列的分析,也就是测定基因在多个时间点的表达值,通过聚类和主成分分析等手段寻找共表达基因;二是基因表达差异的显著性分析;三是蛋白质调控功能的研究.另一方面,是基于生物网络的聚类,如蛋白质相互作用网络的分析等.本研究结合生物大数据的研究背景,综合概述常用的聚类方法在生物数据分析中的应用.
2 聚类分析
2.1聚类分析概念
聚类是现阶段计算机科学领域最热门的研究课题之一,在数据挖掘、生物信息学、图像处理、复杂网络(如社交网络)等方面得到了成功应用.对生物大数据的分析和预测是生物信息学的两个重要任务,聚类分析便是该领域中一个非常活跃的研究课题.聚类与分类不同,它不依赖于预先定义的类和带标号的训练实例,也不确定会产生几个不同的簇.
聚类分析作为一种探索性的数据分析方法[16-18],根据所要研究对象(个体)的属性值特征,采用合适的计算方法对其进行整理,将相似度较高的对象划分为一类;再对同一类内个体的共性及不同类间个体的差异性作进一步归纳,从而得出新的规律.聚类方法建立在多元统计基础之上,是模式识别、数据挖掘等领域中常用的基础方法,尤其适用于分析模式类别数目不确定的情况.合理运用聚类分析方法,将复杂生物数据根据某些相似性度量规则(如基于欧式距离的相似矩阵)进行有效挖掘,对于相似基因表达模块、蛋白质功能组等生物内容的研究大有裨益.
在生物信息学中,可以针对不同的数据集进行聚类分析.通过基因表达谱的聚类[19],分析聚在一个簇中的基因,有助于找到表达模式相近的共表达基因组、探索未知基因的功能、研究基因的调控以及细胞分化的过程等,相比分析单个基因其可靠性更高.通过基于网络的聚类分析,可以发现功能相近的家族蛋白,充分了解细胞内部的工作机理.基于网络的数据主要指蛋白质相互作用网络、基因调控网络和代谢网络三种.
传统的聚类方法包括基于划分的聚类和基于层次的聚类.划分聚类包括K-均值聚类、K-中心点聚类等;层次聚类包括基于距离的分层聚类和基于概率的分层聚类等.近年来,生物技术的突飞猛进和大数据的积累,产生了很多新型的聚类方法[20],如谱聚类、自组织映射神经网络聚类、双聚类、二次聚类、模糊聚类,以及基于生物大数据的并行聚类方法[21]等,这些聚类方法给大数据时代带来了无限生机.
2.2聚类分析基本要求
聚类分析借助于不同的相似性度量规则,产生了不同的方法.对于不同的数据类型、不同的目的以及不同的应用领域,应该选择合适的聚类方法加以分析.在生物信息发展领域,自1998年Eisen等[22]的基于距离度量的基因表达聚类分析,到2001年Hartmink等[23]的基于调控网络拓扑结构的聚类分析,乃至如今对于大数据的挖掘,都体现了生物数据聚类的发展及挑战.因此,聚类分析在生物领域的应用中也形成了一些基本的要求[24],主要有以下几点.
(1)可伸缩性,指算法除了应用于小规模数据外,还要能够适应大数据背景下的要求,因此要保证算法的时间复杂度足够低,占用空间足够小.
(2)适应性,指算法对于目标数据类型的识别,除了能处理数值型数据(如基因的表达值),还要能处理非数值型数据(如网络类型、时间序列类型、生成树类型等).
(3)可用性.目前数据库中存储的数据可能是任意形状的,因此要求算法具备能够发现任意形状的聚类能力.
(4)弱依赖性.很多聚类方法都要求用户输入一些参数(如聚类数目、支持度等),这些参数的值都可能对聚类分析的结果产生很大影响.
(5)对高维数据分析的能力.传统的数据分析对低维数据效果良好,却不一定适合大数据背景下的高维数据,因此聚类分析应该具有降维分析或直接分析的能力.
3 常用的聚类方法
3.1基于基因表达谱的聚类
3.1.1K-均值聚类
K-均值聚类[25]的中心用各类别中所有数据的平均值表示,首先选k个初始点放入簇中计算平均值,然后重新将每个点放入与平均值距离最近的簇,重新计算平均值,直到均值不变或变化小于某一阈值.因此,这种方法一般被称为基于“质心”的技术.需注意的是,在一般情况下,这些质心不是X中的点,虽然它们属于同一个空间.K均值常常被称为Lloyd's算法,其目标是在尽量减小簇内误差平方和的基础上选择质心,即一组N个样本集合X分为不相交的K个簇,各由簇中的样本均值µj表示:

K-均值聚类的过程[26]如图2所示.

图2 K-均值聚类的过程Fig.2 Process of K-means clustering
2014年,Marco等[27]的基于分岔理论的单细胞分析聚类实验中,从小鼠胚胎早期阶段分离出438个体细胞,根据其基因的表达水平,使用高通量的反转录聚合酶链式反应(reverse transcription polymerase chain reaction,RT-PCR)定量选择48个基因,其中包括发育过程中的27个关键转录因子.从单细胞到64细胞阶段,在7个不同的时间点提取细胞,每一个时间点对应一个细胞加倍阶段.对这些数据运用K-均值聚类方法和间距统计方法进行聚类.在每一个时间点,将每个细胞根据其基因表达谱分配给一个父类簇.为了确定某一分岔事件是否发生,将每个父类簇的子簇根据K-均值聚类进一步分为2个不同的簇,并采用间隙统计来选择单簇或双簇模型.重复上述过程,直到最后的时间点.这样就可以创建细胞层次的二叉树初始估计模型.
下面定义描述最佳全局基因表达模式二叉树的结构.通过惩罚似然函数评估每个参数的性能,即

式中,θ表示所有参与定义树结构中的参数;x为观测数据;µc和µa(c)分别为簇c和a(c)的中心,其中a(c)为c的父类簇;λ为事先定义的常量,本实验中设置λ=1.将最终的聚类结果[27]投影到空间上(见图3).

图3 细胞阶段聚类结果Fig. 3 Clustering results of cell stages
图3显示了两个分岔方向上跨平面的整体结构,其中X32和X64不完全正交,每种颜色代表不同的集群,父子集群对由直线连接.图3进一步证明了细胞分化过程中的两个重要阶段,32细胞阶段和64细胞阶段,这对于细胞分化的动力学研究非常有益.
3.1.2K-中心点聚类
K-中心点聚类[28]不同于K-均值聚类,而是选取一个到其他所有点距离之和最小的点作为中心点.这种聚类方法能处理任意类型数据的属性,对异常数据不敏感,因此这些看似异常的数据可能会导致聚类结果的均值变得“扭曲”.与K-均值聚类一样,K-中心点聚类也可以采用欧几里得距离来衡量样本点之间的相似性,终止条件是当所有的类簇的质点都不再发生变化时,认为聚类结束.这里,距离的定义如下:

K-中心点聚类在小型数据集上运行良好,但不适合大数据集,其算法复杂度为O(k(nk)),相对较高.K-中心点聚类的过程[25]如图4所示.
2014年,Trapnell等[29]的关于细胞拟时间动力学分析的研究中,提出对单细胞进行聚类,将具有相似基因表达的基因聚类为一组.该实验首先将具有相似表达趋势的基因分为一组,因为这些细胞可能共享某些生物学功能和调控.一旦每个基因具有广义相加模型(generalized additive models,GAM)值,这些模型就可以用来预测拟时间功能的平滑响应曲线.规范这些曲线,允许所有基因的数据跨拟时间设置有效的K-中心点聚类.根据方向和时间,基因x和y之间的成对距离为

式中,ρx,y表示响应曲线的Pearson相关系数,簇对应的基因遵循相同的相对动力学趋势.在实验中,聚类对象不是原始数据,而是基于GAM响应曲线.相对于通常采用的中心点,这种聚类方法具有较小的均方根误差,更符合动力学原理;同时,对于模式分析的支持也更多样化.在这项研究中,聚类分析了全部可检测的基因表达,无需考虑拟时间序列调控的意义.K-中心点聚类在基因的log转换和标准化后对基因拟时间的GAM进行聚类.聚类应用R语言中的PAM包进行.实验产生了6个区别明显的簇,并最终根据聚类结果对6个簇进行顺式调控分析.

图4 K-中心点聚类的过程Fig.4 Process of K-mediods clustering
3.1.3层次聚类
层次聚类[25]是一种将数据结构化为组群对象,通过自上而下(或自下而上)的迭代,构建相似嵌套集群的聚类方法.层次聚类的结构常用树状图来表示,树根即为所选样本的集合,叶子节点是单一的样本数据.自下而上(凝聚)的聚类[20]是由每个单一样本构成初始簇,根据欧式距离(见式(3))或余弦距离等度量标准,逐层合并相似簇;自上而下(分裂)的聚类是由所有样本构成初始簇,逐层分裂为相似度较低的集合.层次聚类还衍生出了基于密度和基于网格[30]等的聚类方法,特别适用于中等规模的数据集,具有简单、直接的优点.自下而上的层次聚类过程如图5所示.
2015年,Levine等[31]的关于数据驱动下急性骨髓白血病的表现型研究中,对16个小儿急性髓细胞白血病(acute myelocytic leukemia,AML)患者以及5个正常人总共1.5亿个高维数据定义的表型细胞作关联性分析.对细胞表面的16个信号特征和内部的14个抗体信号特征进行观察和数据采样,以Jaccard系数作为相似度的衡量标准,对一组干细胞和相关分化细胞进行分层聚类,这些病人的层次聚类描述显示一些遗传异质性与遗传标志物相关,细胞表面标志物并不能作为细胞表型变化的衡量标准.因此,在研究细胞变化的过程中,通过细胞内的信号分泌物才能得到比较准确的信息.

图5 自下而上的层次聚类过程Fig.5 Process of bottom-up hierarchical clustering
随着生物技术的逐渐成熟,大量生物数据不断产生,数据维度也越来越高.在一般情况下,对高维数据的聚类主要分为子空间聚类法和维归约法.子空间聚类法又大致分为子空间搜索法、基于相关性聚类法和双聚类法.双聚类[32]是指通过二维空间上两个维度同时聚类的方法,比如在基因表达上的应用中,通常所说的基因表达数据或DNA微阵列数据是一个基因-样本/条件矩阵,其中每行表示一个基因,每列表示一个样本或条件,从聚类的角度看即是对两个维度的分析.2012年,Gerstein等[33]的关于人类调控网络的DNA元件百科全书(encyclopedia of DNA elements,ENCODE)数据研究中,通过双聚类对基因表达谱进行分析,最终发现了特定背景下几种共关联的表达基因.当然,面对高维大数据也可以采用谱聚类等方法,或者借助于MapReduce工具来实现.
3.2基于蛋白质网络的聚类
分子生物学是一门研究生物本身组成成分的科学,主要包括核酸、蛋白质和酶的结构和合成、功能和代谢、表达和调控以及它们之间的相互作用等,以此来揭示生命的本质,是目前生物发展进程中的基础学科.而研究蛋白质的相互作用[34](protein-protein interaction,PPI)则是生命科学研究中的重要一环.蛋白质作为生命功能的执行者,尤其是随着后基因组时代的到来,蛋白质相互作用网络的研究更是成为系统生物学研究中的重要内容之一.比较成熟的研究理论认为,蛋白质并不是单独发挥作用的,而是通过与相关蛋白质的相互作用,在空间和时间上协调一致,共同调控、维持细胞的特定功能.通过对蛋白质相互作用网络的聚类分析,可以找到感兴趣的蛋白质功能组,这对于生物体的行为研究、未知蛋白功能的预测以及药物的设计都具有重要的作用.
利用聚类方法获取蛋白质网络中的生物相关功能模块是目前的研究热点之一[35].根据采取方式的不同,主要有基于图论的方法(graph-theoretic approach)、基于模拟流的方法(flow simulation-based approaches)、基于谱聚类的方法(spectral clustering-based approaches)、基于监督学习的方法(supervised clustering approaches)、基于依赖核心的方法(core attachmentbased approaches)、基于群体智能的方法(swarm intelligence-based approaches)等.具体划分[36]如表1所示.

表1 蛋白质网络聚类方法分类Table 1 Classification of protein network clustering
2008年,Blondel等[37]基于层次聚类和模块性最优化提出了Louvain方法.该方法实现了在模块性最优化的基础上,快速得到网络分层的效果.此后,Xie等[38]将Louvain方法应用在蛋白质模块比对的可视化分析中,利用Louvain层次化的网络聚类结果,用可视化形式分析生物分子网络比对的结果.层次聚类之后的网络能很好地以整体和局部两种视图分析生物分子网络的特点(见图6).同时,结合蛋白质功能模块与蛋白质相互作用网络的比对结果,有助于发现生物相关功能模块的比对结果.为便于进行相关功能蛋白的研究,还可以使用主成分分析(principal component analysis,PCA)方法[39].

图6 Louvain层次化的蛋白质网络聚类可视化效果[38]Fig.6 Visualization effect of Louvain hierarchical clustering protein network[38]
4 实验对比
众所周知,基因表达数据的聚类分析为生物研究提供了强有力的技术支持,但是不同的聚类方法却会导致不同的实验结果.本研究对GEO中小鼠胚胎成纤维细胞体外低温下基因表达差异性[40]数据进行聚类,数据由Affymetrix公司提供[41],总共包括不同时间点的13组样本数据.这种基于时间序列[42]的聚类可广泛应用于不同领域,并且已被证明是非常有效的.将胚胎成纤维细胞体外暴露在轻度低温(32°C)环境或常温(37°C)环境下长达18 h,在不同时间点对数据进行采样.低温是一种临床上有效的治疗各种缺氧和缺血的手段之一.37和32°C取样时间点分别为0,0.5,1.0,2.0,4.0,8.0,18.0 h和0.5,1.0,2.0,4.0,8.0,18.0 h,这些样本分别编号为GSM1310500~GSM1310512,共计13组.正常温度下样本包括GSM1310500~GSM1310506,低温下样本包括GSM1310507~GSM1310512.聚类方法采用K-均值、K-中心点和分层聚类的方法,相似性度量选择欧氏距离,初始选择k=4.以热图(heatmap)的形式展现部分聚类结果如图7~9所示[41].
实验聚类的具体数据可参照文献[41],图7~9中颜色接近程度表示基因之间的相似表达程度.根据不同时间点的基因聚类结果可以得出同一时间点的共表达基因组,这为细胞分化的阶段性分析提供了强有力的证据.
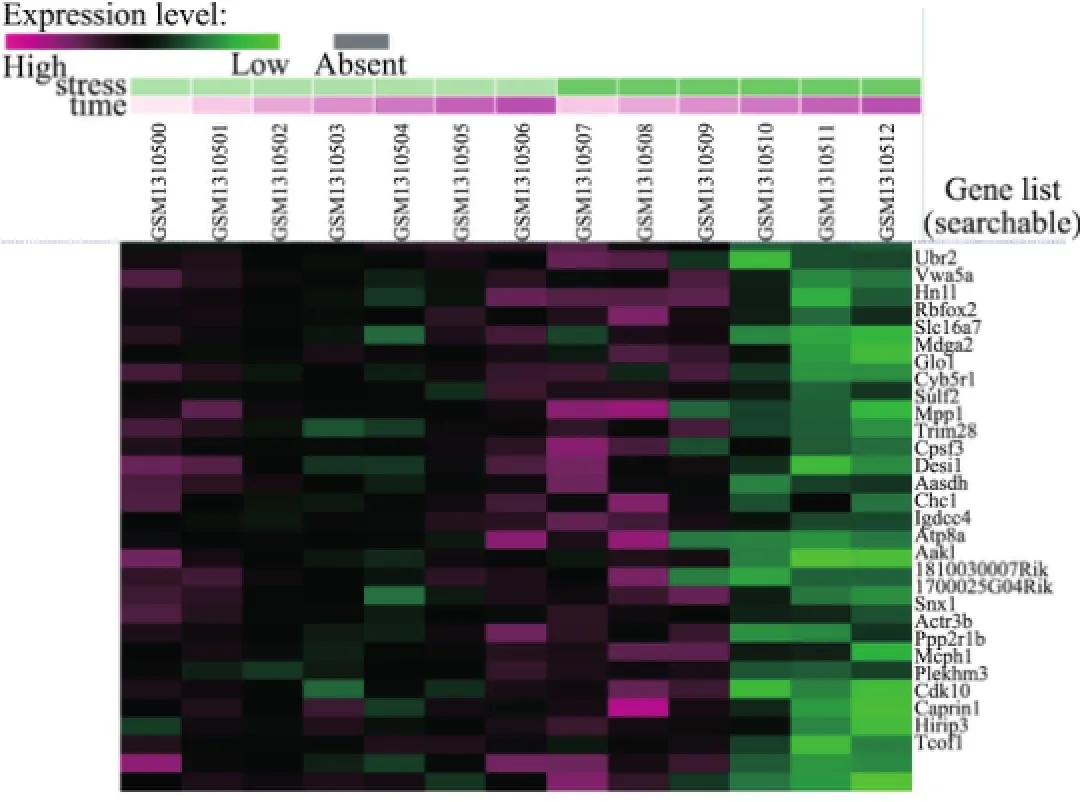
图7 K-均值聚类结果Fig.7 Results of K-means clustering

图8 K-中心点聚类结果Fig.8 Results of K-mediods clustering

图9 层次聚类结果Fig.9 Results of hierarchical clustering

表2 三种聚类方法结果Table 2 Results of the three clustering methods
从3组聚类结果的基因表达谱可以看出:①从整体上而言,正常温度下基因的表达值相对较高;②某些基因在低温下0.5~1.5 h时表达增强.表2列出了三种聚类方法的结果,通过对比可以发现,K-均值和K-中心点聚类结果显示一致,却与层次聚类结果有所差异.也就是说,当采用不同的聚类方法时,得到的聚类结果将受到影响.表2中三种算法都聚在一个簇的样本分别以下划线、星号(*)、井号(#)及梅花符号(♣)标记,这些样本的聚类具有更高的可信度,也是在进一步分析中需要着重关注的内容.
5 结束语
生物信息学是一个成长中的跨学科领域[43-44],生物数据日新月异、逐年增长.聚类是数据挖掘的基本方法之一,在基因表达和生物网络等数据挖掘过程中起到了举足轻重的作用.后基因组时代伴随着二代甚至三代测序技术的发展,单一的聚类方法已经无法满足当前的大数据要求.另外,高噪声数据的干扰也是亟待解决的问题.综上所述,选择和结合不同的聚类方法,针对不同的生物数据进行综合分析与比较,进一步提出新的适用于大数据挖掘的方法,将有助于揭晓更多的生物奥秘.
[1]赵屹,谷瑞升,杜生明.生物信息学研究现状及发展趋势[J].医学信息学杂志,2012,33(5):2-6.
[2]KOBOLDT D C,STEINBERG K M,LARSON D E,et al.The next-generation sequencing revolution and its impact on genomics[J].Cell,2013,155(1):27-38.
[3]任艳姣.生物信息学数据整合的应用研究[D].长春:吉林大学,2012.
[4]BENSON D A,KARSCH-MIZRACHI I,LIPMAN D J,et al.GenBank[J].Nucleic Acids Research,2000,28(1):15-18.
[5]UETZ P,ETZOLD T.The EMBL/EBI reptile database[J].Herpetological Review,1996,27(4):174-175.
[6]BARRETT T,WILHITE S E,LEDOUx P,et al.NCBI GEO:archive for functional genomics data sets-update[J].Nucleic Acids Res,2013,41:D1005-D1010.
[7]王洪昌,丁立军,黄宇.生物信息学中模式识别技术应用与发展[J].医学信息学杂志,2013(11):7-10.
[8]LI Y,CHEN L.Big biological data:challenges and opportunities[J].Genomics,Proteomics and Bioinformatics,2014,12(5):187-189.
[9]MARx V.Biology:the big challenges of big data[J].Nature,2013,498(7453):255-260.
[10]SCHUSTER S C.Next-generation sequencing transforms today's biology[J].Nature,2007,200(8):16-18.
[11]REIS-FILHO J S.Next-generation sequencing[J].Breast Cancer Res,2009,11(S3):S12.
[12]MARCOTTE E M,DATE S V.Exploiting big biology:integrating large-scale biological data for function inference[J].Briefings in Bioinformatics,2001,2(4):363-374.
[13]ARONOVA E,BAKER K S,ORESKES N.Big science and big data in biology:from the international geophysical year through the International Biological Program to the Long Term Ecological Research(LTER)Network,1957—present[J].Historical Studies in the Natural Sciences,2010,40(2):183-224.
[14]MADEIRA S C,OLIVEIRA A L.Biclustering algorithms for biological data analysis:a survey[J]. IEEE/ACM Transactions on Computational Biology and Bioinformatics(TCBB),2004,1(1):24-45.
[15]杨春梅,万柏坤,高晓峰.基因表达聚类分析技术的现状与发展[J].生物化学与生物物理进展,2003,30(6):974-979.
[16]黄金.聚类和分类技术在生物信息学中的应用[D].哈尔滨:黑龙江大学,2005.
[17]陈亮.聚类算法及其在生物信息学中的应用[D].无锡:江南大学,2012.
[18]REDDY C K,AL HASAN M,ZAKI M J.Clustering biological data[M]//Data clustering:algorithms and applications.London:Chapman and Hall/CRC,2013:381-414.
[19]ERCIYES K.Clustering of biological sequences[M]//ERCIYES K.Distributed and sequential algorithms for bioinformatics.Berlin:Springer International Publishing,2015:135-160.
[20]AGGARWAL C C,REDDY C K.Data clustering:algorithms and applications[M].Boca Raton:CRC Press,2014.
[21]WANG M,ZHANG W,DING W,et al.Parallel clustering algorithm for large-scale biological data sets[J].PLoS ONE,2014,9(4):e91315.
[22]EISEN M B,SPELLMAN P T,BROWN P O,et al.Cluster analysis and display of genome-wide expression patterns[J].Proceedings of the National Academy of Sciences,1998,95(25):14863-14868.
[23]HARTEMINK A J,GIFFORD D K,JAAKKOLA T,et al.Using graphical models and genomic expression data to statistically validate models of genetic regulatory networks[C]//Pacific Symposium on Biocomputing.2001:422-433.
[24]苏志中.聚类分析研究及其在生物数据分析中的应用[D].长沙:湖南大学,2009.
[25]周洋.基因表达谱数据聚类分析的研究[D].咸阳:西北农林科技大学,2014.
[26]HAN J,KAMBER M,PEI J.Data mining:concepts and techniques:concepts and techniques[M]. Amsterdam:Elsevier,2011.
[27]MARCO E,KARP R L,GUO G,et al.Bifurcation analysis of single-cell gene expression data reveals epigenetic landscape[J].Proceedings of the National Academy of Sciences,2014,111(52):E5643-E5650.
[28]张琛.生物信息学中的基因表达谱数据分析研究[D].长春:吉林大学,2008.
[29]TRAPNELL C,CACCHIARELLI D,GRIMSBY J,et al.The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells[J].Nature Biotechnology,2014,32(4):381-386.
[30]MURTAGH F,CONTRERAS P.Algorithms for hierarchical clustering:an overview[J].Wiley Interdisciplinary Reviews:Data Mining and Knowledge Discovery,2012,2(1):86-97.
[31]LEVINE J H,SIMONDS E F,BENDALL S C,et al.Data-driven phenotypic dissection of AML reveals progenitor-like cells that correlate with prognosis[J].Cell,2015,162(1):184-197.
[32]安平.基因表达数据的双聚类分析方法研究[D].苏州:苏州大学,2013.
[33]GERSTEIN M B,KUNDAjE A,HARIHARAN M,et al.Architecture of the human regulatory network derived from ENCODE data[J].Nature,2012,489(7414):91-100.
[34]王正华,董蕴源,王勇献.蛋白质相互作用网络的几种聚类方法综述[J].国防科技大学学报,2009,31(004):81-86.
[35]刘昊,廖波,彭利红.基于蛋白质相互作用网络的聚类算法研究[J].计算机工程与应用,2009,44(30):142-144.
[36]JI J Z,ZHANG A D,LIU C N,et al.Survey:functional module detection from protein-protein interaction networks[J].IEEE Transactions on Knowledge and Data Engineering,2014,26(2):261-277.
[37]BLONDEL V D,GUILLAUME J L,LAMBIOTTE R,et al.Fast unfolding of communities in large networks[J].Journal of Statistical Mechanics:Theory and Experiment,2008,DOI:10.1088/1742-5468/2008/10/P10008.
[38]XIANG C J,XIE J,GU Y L,et al.Visualization of module alignment discovery[C]//Control Conference(CCC).2015:8545-8549.
[39]ASUR S,UCAR D,PARTHASARATHY S.An ensemble framework for clustering protein-protein interaction networks[J].Bioinformatics,2007,23(13):i29-i40.
[40]TIBSHIRANI R,HASTIE T,EISEN M,et al.Clustering methods for the analysis of DNA microarray data[R].Stanford:Stanford University,1999.
[41]STEN I,ANSGAR H C,RIIN R,et al.Estimating differential expression from multiple indicators[J].Nucleic Acids Research,2014,42(8):e72.
[42]LIAO T W.Clustering of time series data—a survey[J].Pattern Recognition,2005,38(11):1857-1874.
[43]TORARINSSON E,HAVGAARD J H,GORODKIN J.Multiple structural alignment and clustering of RNA sequences[J].Bioinformatics,2007,23(8):926-932.
[44]FITZGERALD P C,SHLYAKHTENKO A,MIR A A,et al.Clustering of DNA sequences in human promoters[J].Genome Research,2004,14(8):1562-1574.
Survey of clustering methods for big data in biology
LU Dongfang,XU Junfu,XIANG Chaojuan,XIE Jiang
(School of Computer Engineering and Science,Shanghai University,Shanghai 200444,China)
With the implementation of the Human Genome Project and the rapid development of biological experiment technology,biological data sharply grow and continuous accumulate.Age of big data in biology is coming.In the post genomic era,single statistical models are gradually replaced with combination of intelligent and comprehensive analyses. Clustering is the core of data mining.This paper describes the state-of-the-art technology of big data in bioinformatics,and summarizes several popular clustering methods on gene expression profiling and biological networks.Furthermore,some experiments are made to compare different clustering methods on the time series data of mouse embryonic fibroblasts,showing that different clustering methods have different results.To achieve more reliable conclusions for highly noisy biological data,it is necessary for investigators to do comprehensive analyses by selecting and combining proper clustering methods.
big data in biology;data analysis;clustering method
TP 39
A
1007-2861(2016)01-0045-13
10.3969/j.issn.1007-2861.2015.04.018
2015-11-30
国家自然科学基金重大研究计划项目(91330116);教育部留学回国人员科研启动基金资助项目
谢江(1971—),女,副教授,博士,研究方向为生物信息学、高性能计算.E-mail:jiangxsh@shu.edu.cn
