青海东方蜜蜂贵德种群的遗传分化地位
2016-09-20陈振华李文靖张同作林恭华中国科学院西北高原生物研究所西宁80002青海中科蜜蜂研究院西宁80003青海青藏华峰中蜂蜂业有限公司贵德8700
赵 芳 陈振华 李文靖 张同作 皮 立 林恭华(中国科学院西北高原生物研究所,西宁8000;2青海中科蜜蜂研究院,西宁8000;3青海青藏华峰中蜂蜂业有限公司,贵德8700)
青海东方蜜蜂贵德种群的遗传分化地位
赵芳1,2陈振华2,3李文靖1,2张同作1皮立1林恭华1,2
(1中国科学院西北高原生物研究所,西宁810001;2青海中科蜜蜂研究院,西宁810001;3青海青藏华峰中蜂蜂业有限公司,贵德811700)
对青海东方蜜蜂贵德种群(QHGD)进行全基因组测序,并结合现有其他地区东方蜜蜂的分子序列信息,分析QHGD种群的遗传分化地位。测序、组装和比对后得到Cytb、tRNAIle~ND2、16S rRNA、EF1-α、COI~COII序列对应的比对文件。单倍型分析显示,QHGD种群的Cytb、EF1-α、COI~COII序列与国内其他种群有重叠,但tRNAIle~ND2、16S rRNA序列则形成特有的单倍型。基于tRNAIle~ND2、16S rRNA、EF1-α合并序列的Network分析显示,QHGD种群与四川九寨(SCJZ)、四川马尔康(SCMEK)等地方种群成辐射状与一个median vector节点(mv1)连接。本研究的结果表明,青海贵德种群是较为独特的一个进化分支单元,需要在未来的保护和开发利用过程中予以关注。
东方蜜蜂;贵德种群;遗传分化;基因组
青海东方蜜蜂是优良的中华蜜蜂地方品种之一,主要分布于青海省东部农业区。青海东方蜜蜂在分类上属于东方蜜蜂阿坝亚种(Apis cerana abansis)[1],在形态和行为等方面都表现出明显的高海拔、低气温的适应特征[2,3],对青藏高原地区养蜂业发展和生态平衡维持具有重要作用。长期以来,青海东方蜜蜂的养殖多是农户自发的散养模式,没有形成产业化。由于繁育体系不到位,致使种群数量不断衰减,加上外来西方蜜蜂的威胁,青海东方蜜蜂一度处于频临灭绝的边缘[4,5]。
值得一提的是,当前对青海东方蜜蜂的研究,其对象都是青海省民和县的种群[5],而对其他地区的种群则很少涉及。青海省贵德县蜂农自1979年开始利用当地的野生蜂群探索青海东方蜜蜂的科学繁育技术,并于2010年取得突破。以青海青藏华峰中蜂蜂业有限公司为平台,现已繁育优良生产群(命名为贵德种群)1,500群,生产性能表现优异,其产品获得第44届APIMONDIA国际养蜂大会暨博览会金牌和银牌各一枚[6]。为了更好地保护和利用这一优良品种资源,有必要对其遗传特征进行科学描述。本研究对青海东方蜜蜂贵德种群进行基因组重测序,利用分子生物学研究方法,探讨其遗传分化地位。
1 材料方法
1.1样品采集和高通量测序
蜜蜂样品采自青海青藏华峰中蜂蜂业有限公司养蜂场内的健康蜂群,随机选取东方蜜蜂蜂群2群(QHGD1、QHGD2),用50 ml离心管从蜂箱内采集10只工蜂,放入液氮中固定保存,用干冰运输到北京百迈客生物科技有限公司进行高通量测序。实验流程按照Illumina公司提供的标准protocol执行:每群取3只工蜂样品,分别提取基因组DNA,检测合格后用超声波将DNA片段化;对片段化的DNA进行片段纯化、末端修复、3’端加A、连接测序接头;用琼脂糖凝胶电泳进行片段大小选择,对序列长度为500 bp的片段进行PCR扩增形成测序文库;质检合格的文库用Illumina HiSeqTM 2500进行双向测序(pair-end),计划测序深度为40X。对测序得到的原始测序序列(raw reads)进行数据评估、过滤(去除接头、低质量区域等),最终得到用于生物信息学分析的clean reads。
1.2分子标记序列组装
目前,对国内东方蜜蜂进行遗传分化研究,有3篇文献采样范围较广且给出样品的来源信息[7-9]。将贵德生产群的对应序列和这些研究进行比较,可以得到贵德种群的遗传分化地位。这些研究所涉及的分子标记包括4个线粒体DNA片段(Cytb、tRNAIle~ND2、16S rRNA、COI~COII)和1个核基因片段(EF1-α)。首先,从GenBank中下载已经测出的东方蜜蜂线粒体全基因组(登记号:NC_014295.1)[10],以此为饵,用SOAPaligner软件[11]在clean reads数据库中提取匹配的reads。然后,将这些reads与此线粒体全基因组合并成单个文件,用Geneious软件(Biomatters Limited)中的Assembly功能模块,以此线粒体全基因组为模板将这些reads组装成长片段(scaffold)。结合使用blast+[12]和MEGA[13]两个软件,以文献报道的EF1-α序列(GenBank登记号:KF663570.1)为饵,从东方蜜蜂全基因组序列文件中[14]找到包含 EF1-α序列的 scaffold(A_cerana_Scaffold_0111),并截取EF1-α片段及其侧翼500 bp的片段。以同样方法,用SOAPaligner和Geneious软件组装相应片段。
1.3遗传分化地位分析
从GenBank中下载前面提到的3篇文献中所用序列[7-9],用MEGA软件分别将这些序列和本研究中组装得到的贵德种群序列进行比对,去除未匹配区段,最终得到基于不同序列片段的alignment文件。用DAMBE软件分析变异位点数和单倍型组成情况,分析过程考虑插入/缺失位点。由于田崇浩等[8]涉及2个线粒体DNA片段(tRNAIle~ND2和16S rRNA)和1个核基因片段(EF1-α),涉及的变异位点信息较多,我们将3个片段合并,用DNasp软件生成rdf文件(考虑插入/缺失位点)。用Network软件计算单倍型关系(Median Joining模型),并绘制单倍型网络图。
2 结果与分析
2.1基因组测序和目标片段组装
对两群蜜蜂样品(QHGD1、QHGD2)成功进行基因组测序,分别得到84,519,894和85,325,442条raw reads序列,两样本Q20值(质量值大于等于20的碱基占总碱基数的百分比)都在90%和以上。经过滤后分别得到81,629,314和84,873,218条clean reads,以韩国东方蜜蜂[14]为参考基因组,两样本mapped指数(定位到参考基因组的clean reads数占所有clean reads数的百分比)都高于78%,平均测序深度都高于40X,Coverage_ratio_5X值(5X深度及以上的碱基数占参考基因组总碱基数的比例)都高于90%(见表1)。以上结果显示,两份基因组测序质量较好,可以满足序列分析要求。
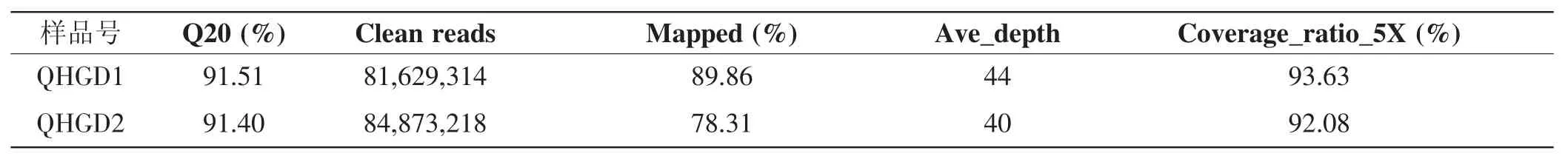
表1 青海东方蜜蜂贵德生产群基因组测序基本情况统计
SOAPaligner软件比对显示,QHGD1和QHGD2分别有939,283和716,069条clean reads与参考线粒体基因组匹配。Geneious软件统计显示,平均碱基覆盖度分别为7,445X和5,677X,经组装后分别得到15,465 bp和15,462 bp长度的单条scaffold,约占参考线粒体基因组长度(15,897 bp)的97%。同时,QHGD1和QHGD2分别有444和361条clean reads与参考EF1-α及其侧翼500 bp片段匹配;平均碱基覆盖度分别为43X和35X,经组装后分别得到1,304 bp和1,286 bp长度的单条scaffold,占参考EF1-α及其侧翼500 bp片段长度(1,306bp)的98%以上。

图1 四川九寨(SCJZ,GenBank No:KF663582.1)群和青海贵德群(QHGD)的tRNAIle~ND2序列比对结果

图2 四川九寨(SCJZ,GenBank No:KF572602.1)群和青海贵德群(QHGD)16S rRNA序列比对结果
2.2序列比对和遗传分化地位分析
将本研究得到的线粒体或EF1-α及其侧翼片段scaffold分别与已经发布的 Cytb、tRNAIle~ND2、16S rRNA、EF1-α、COI~COII序列合并,比对后截取同源序列,最终得到5个alignment文件。DAMBE软件的分析显示,QHGD1和QHGD2两个群的序列在上述5个基因区段完全相同,为叙述方便,将两者视为同一个基因序列对待,统称为QHGD(即“青海贵德”4个字的拼音首字母缩写)序列。
基于Cytb序列的分析显示,QHGD序列与甘肃天水(GSTS,EF180093.1)、福建福州(FJFZ,FJ229471.1)、四川宜宾(SCYB,FJ229473.1)、山西沁源(SXQY,FJ229480.1)种群的序列[7]完全一致。基于tRNAIle~ND2、16S rRNA序列的分析显示,QHGD序列与现有的所有其他19种群[8]都不相同(图1~2)。基于EF1-α序列的分析显示,QHGD序列与绝大多数种群(JLAT、JLHD、JLHN、SDFX、SXZQ、SXTG、SXQY、SXQS、GSTS、SCMEK、YNBS、YNKM、YNXSBN、GXQZ、FJFZ、HNHK,KF663570.1)所含有的单倍型[8]完全相同。基于COI~COII序列的分析显示,QHGD序列与 Japan1 (AB078733.1)单倍型[9]完全相同。
基于tRNAIle~ND2、16S rRNA、EF1-α合并序列的Network分析显示,QHGD种群与国内其他19个地方种群没有直接交集。相反,QHGD与四川九寨(SCJZ)、四川马尔康(SCMEK)、山西左权(SXZQ)、山西沁源(SXQY)、山东费县(SDFX)共同指向一个中值向量(median vector)节点mv1(图3),即这些种群为同一个祖先节点辐射状分化而成。

图3 基于tRNAIle~ND2、16S rRNA、EF1-α合并序列的Network关系图
绿色数字表示两两节点间的差异位点数,未标注的表示1个位点差异;红色字母QHGD表示青海贵德生产群。字母代码所表示的地点信息与田崇浩(2014)一致:JLAT,吉林安图;JLHD,吉林桦甸;JLHN,吉林辉南;SDFX,山东费县;SXZQ,山西左权;SXTG,山西太谷;SXQY,山西沁源;SXQS,山西沁水;GSTS,甘肃天水;SCJZ,四川九寨;XZMEK,四川马尔康;SCYB,四川宜宾;YNBS,云南保山;YNKM,云南昆明;YNXSBN,云南西双版纳;GDGZ,广东广州;GXQZ,广西钦州;FJFZ,福建福州;HNHK,海南海口。
3 讨论
由于地处偏僻和产业化水平较低等原因,长期以来,学界对青海地区的东方蜜蜂种群关注较少。杨冠煌等[1]首先提出,青海东部地区的东方蜜蜂属于阿坝亚种,这一说法在《中国畜禽遗传资源志·蜜蜂志》中也得到体现。然而这一论述是基于青海省和四川阿坝地区地理距离和地貌特征相近而进行的推测,并无直接研究证据支持。李华等[2]基于形态测量数据的研究表明,青海民和地区的东方蜜蜂与阿坝九寨种群有明显不同。本研究的结果显示,QHGD与阿坝地区的SCJZ和SCMEK种群从同一个节点辐射状分化形成不同的进化枝,换句话说,青海贵德和阿坝地区的东方蜜蜂种群在遗传上并无隶属关系。以SCJZ为例,青海贵德种群在tRNAIle~ND2和16S rRNA序列上与其分别具有4 和2个位点差异(图1~2)。此外,从Network关系上看,SCJZ和SCMEK与mv1节点仅有1个位点差异,而QHGD种群则与mv1节点间的位点差异达6个之多。以上结果表明,青海贵德种群可能不属于阿坝亚种,相反,是较为独特的一个进化分支单元。
Tan et al[9]是目前已发表的唯一涉及青海东方蜜蜂(民和种群)的报道。其研究结果显示,在COI~COII序列上,青海民和种群的单倍型(Qinghai,GenBank No:KT174438)和四川的部分种群单倍型相同。有趣的是,本研究中青海贵德的COI~COII序列与Tan et al[9]文中的 Japan1(AB078733) 完全相同,与 Qinghai (KT174438)反而有1个碱基位点的差异。这是否意味着青海民和和贵德两个东方蜜蜂种群有本质上的差异,有待进一步研究。
由于各种条件的限制,东方蜜蜂遗传分化的研究有些混乱。形态学研究过程中,会带入相对较多的主观意见,而结合分子生物学研究手段将大大降低这种主观性[15]。当前,绝大多数涉及东方蜜蜂遗传分化的研究,仅使用1个或少数几个序列片段,由于缺乏足够的变异信息,不同地理种群之间单倍型交叠,导致结果不明朗。以本研究为例,青海贵德种群在3个基因序列(Cytb、EF1-α、COI~COII)上都分别与多个其他地理种群有单倍型重叠,无法据此确定彼此的遗传分化地位。相反,由于tRNAIle~ND2、16S rRNA、EF1-α属于同一批样品,将其合并后分析,可以得到相对明晰的结论,足见多个基因标记方法的优越性。本研究对青海东方蜜蜂贵德种群进行基因组测序,得到巨量的序列信息,可以与现有其他地区的遗传分化研究数据做到较好的对接,为阐明其遗传分化地位并服务于其保护和利用提供重要的科学依据。
[1]杨冠煌,许少玉,匡邦郁,等.东方蜜蜂Apis cerana Fab.在我国的分布及其亚种分化 [J].云南农业大学学报,1986,1(1)∶89-92.
[2]李华,张祖芸,谭垦.青海东方蜜蜂的形态学研究 [J].蜜蜂杂志,2008,12∶5-6.
[3]李良德,马少荣,白斌.青海东方蜜蜂遗传资源调查 [J].中国蜂业,2011,62(1)∶26-27.
[4]张学功,张学成,马少荣.青海中蜂养殖现状及保护措施[J].中国蜂业,2010,6(9)∶34-35.
[5]冶兆平.再议青海东方蜜蜂养殖现状与保护措施 [J].中国蜂业,2014,65∶64-66.
[6]王建梅,陈黎红,胡玭玭.中国养蜂学会蜂业代表团在第44 届APIMONDIA国际养蜂大会暨博览会荣获多项奖牌[J].蜜蜂杂志,2015,11∶45.
[7]高鹏飞,赵慧婷,张春香,等.基于线粒体Cyt b基因部分序列的中国东方蜜蜂不同地理种群的系统发育 [J].动物学报,2008,54(6)∶1005-1013.
[8]田崇浩.中国境内东方蜜蜂群体亚分化研究 [D].山西农业大学硕士学位论文,2014.
[9]Tan K,Qu Y,Wang Z,et al.Haplotype diversity and genetic similarity among populations of the Eastern honey bee from Himalaya-Southwest China and Nepal(Hymenoptera∶Apidae)[J].Apidologie,2015,DOI∶10.1007/s13592-015-0390-x.
[10]Tan HW,Liu GH,Dong X,et al.The complete mitochondrial genome of the Asiatic cavity-nesting honeybee Apis cerana(Hymenoptera∶Apidae)[J].PLoS One,2011,6(8)∶e23008.
[11]Gu S,Fang L,Xu X.Using SOAPaligner for short reads alignment[J].Current Protocols in Bioinformatics,2013,44∶11.11.1-11.11.17.
[12]Zhang Z,Schwartz S,Wagner L,et al.A greedy algorithm for aligning DNA sequences[J].Journal of Computational Biology,2000,7(1-2)∶203-214.
[13]Tamura K,Stecher G,Peterson D,et al.MEGA6∶molecular evolutionary genetics analysis version 6.0[J].Molecular Biology and Evolution,2013,30∶2725-2729.
[14]Park D,Jung JW,Choi BS,et al.Uncovering the novel characteristics of Asian honey bee,Apiscerana,by whole genome sequencing[J].BMC Genomics,2015,16∶1.
[15]杨洁,和绍禹,苗永旺,等.东方蜜蜂(Apis cerana)的分类研究进展.蜜蜂杂志,2011,3∶13-15.
Genetic variation status of Apis cerana population from Guide,Qinghai
Zhao Fang1,2Chen Zhenhua2,3Li Wenjing1,2Zhang Tongzuo1Pi Li1Lin Gonghua1,2
(1 Northwest Institute of Plateau Biology,Chinese Academy of Sciences,Xining 810001;2 Qinghai-CAS Institute of Apicultural Research,Xining 810001;3 Qinghai Qingzang-Huafeng Eastern Honeybee Industry Co.,Ltd,Guide 811700)
We sequenced the wholegenome of Apis cerana from Guide population(QHGD)and,combining with sequence data on A.cerana populations from other regions,we analyzed the genetic variation status of the QHGD population.After sequencing,assembling,and aligning,we obtained alignments for five DNA fragments including Cytb,tRNAIle~ND2,16S rRNA,EF1-α,and COI~COII.Haplotype analysis showed that the QHGD population shared haplotypes with other populations on Cytb,EF1-α and COI~COII,in contrast,the QHGD population formed a unique haplotype on tRNAIle~ND2 and 16S rRNA,respectively.Network analysis based on a combination of tRNAIle~ND2,16S rRNA and EF1-αshowed that the populations of QHGD,SCJZ,SCMEK,etc.formed a radialshape from a median vector (mv1).Our study indicated that the QHGD population was a very unique genetic branch,and people should pay attention to their special nature in the future conservation and developing processes.
Apis cerana;Guide population;genetic variation;genome
中国科学院青年创新促进会人才项目(NO.2015352)
林恭华,副研究员,硕导,主要从事青藏高原动物生态学研究,E-mail:lingonghua@nwipb.cas.cn
