两种Zn(Ⅱ)配合物电子结构和光谱性质的理论研究
2016-08-16李永红
汪 欣,李永红
(河南大学 化学化工学院,环境与分析科学研究所,河南 开封 475004)
两种Zn(Ⅱ)配合物电子结构和光谱性质的理论研究
汪欣,李永红*
(河南大学 化学化工学院,环境与分析科学研究所,河南 开封 475004)
为了探究Zn(Ⅱ)配合物Zn(ATSM) (A)和Zn(BTSC)(DMSO) (B)的电子结构和光谱性质,采用M06方法优化了它们的基态几何构型,并利用计算得到的电子结构信息绘制了配合物在吸收过程中的电子云分布图.理论模拟出的吸收光谱数据与实验结果吻合较好.而且,在理论上检测到了实验上没有报道到的吸收峰.
密度泛函理论; 吸收光谱; Zn(Ⅱ)配合物
氧气对于所有的生物组织来说都是必不可少的.它几乎可以影响所有细胞的生长周期,甚至包括固体肿瘤细胞[1].许多常见疾病的一个共同特点是生物组织的氧化作用减弱,这种现象也称为缺氧[2-4].研究表明,缺氧会降低放射的效果,从而将大大提高癌症病人的死亡率[5].及早识别出细胞组织缺氧的位置和缺氧的程度对于临床上有效控制这类疾病是至关重要的[3].因此,大量的研究都致力于设计新型的显像剂从而能高效、可靠地应用于相关疾病治疗中去[6-7].金属配合物常被用于活细胞成像.其中铜配合物,例如Cuprum biacetylbis(4-methyl-3-thiosemicarbazone) [Cu(ATSM)]及其衍生物多年来一直是人们研究的对象[8].然而,通过简单的荧光显微镜很难观察到铜配合物的细胞毒性和它们的亚细胞行为.近年来,相应的金属锌配合物也被作为一种性能较好的显像剂应用于活细胞显像中.
COWLEY等[9]合成了一种Zn(Ⅱ)配合物Zinc biacetylbis(4-methyl-3-thiosemicarbazone) [Zn(ATSM)](A).MARTIN等采用间略微分重叠方法(ZINDO)和含时密度泛函理论(TD-DFT)计算了它的结构和紫外-可见光谱信息[10]. 根据配合物A的制备过程,DAYAL等[11]合成了另外一种Zn(Ⅱ)配合物Zinc benzilbis(4-pyrrolidinyl-3-thiosemicarbazone) [Zn(BTSC)(DMSO)](B).本文采用密度泛函理论(DFT)分别优化了配合物A和B的基态几何构型.接着,利用含时密度泛函理论模拟了它们的吸收光谱性质.我们的研究目标是通过分析这两种配合物的详细电子结构和轨道跃迁情况来阐明它们的吸收光谱性质.其中,配合物B的电子跃迁信息在理论上从未被报道过.我们希望该项工作可以帮助人们更好地理解缩氨基硫脲类金属配合物的光学性能.
1 计算方法
采用M06[12]方法优化配合物A和B的基态几何构型.非金属原子(H、C、N、O和S)采用6-31+G(d)基组[13],Zn原子采用LANL2DZ赝势基组[14].在相同水平下计算了驻点的振动频率.没有虚频存在意味着每种构型均为势能面上的最小点.在基态优化构型基础上,采用TD-M06方法[15]并结合极化连续介质模型(PCM)[16-17]计算了目标配合物在二甲亚砜溶剂中的电子结构和紫外吸收光谱.所有的计算都采用Gaussian09[18]软件包完成的.
2 结果和讨论
2.1几何构型
在M06/6-31+G(d)-LANL2DZ水平下优化了配合物A和B的几何结构.图1和2分别为配合物A和B的球棍模型和结构简图.表1列出了优化得到的键长、键角参数及相关的实验数据.计算结果表明,理论值和实验值之间的最大误差为0.008 nm,这说明了采用M06方法优化配合物A和B的基态构型具有一定的可靠性.
DAYAL等[11]通过紫外、红外、质谱、核磁共振氢谱及碳谱等手段对配合物A进行表征,发现配合物A中的锌原子与配体ATSMH2(biacetylbis(4-methyl-3-thiosemicarbazone))中-HC=N-基团的氮原子及-C=S基团中的硫原子进行配位形成了一个四配位的结构.配合物B中,配体BTSCH2(benzilbis(4-pyrrolidinyl-3-thiosemicarbazone))提供4个配位原子,即-HC=N-基团中的2个氮原子和-C=S基团中的2个硫原子与锌原子进行配位.第5个配位原子来自于溶剂二甲亚砜(DMSO)中-S=O基团的氧原子.这些配位原子围绕着中心金属锌原子形成了一个扭曲的四方锥结构.
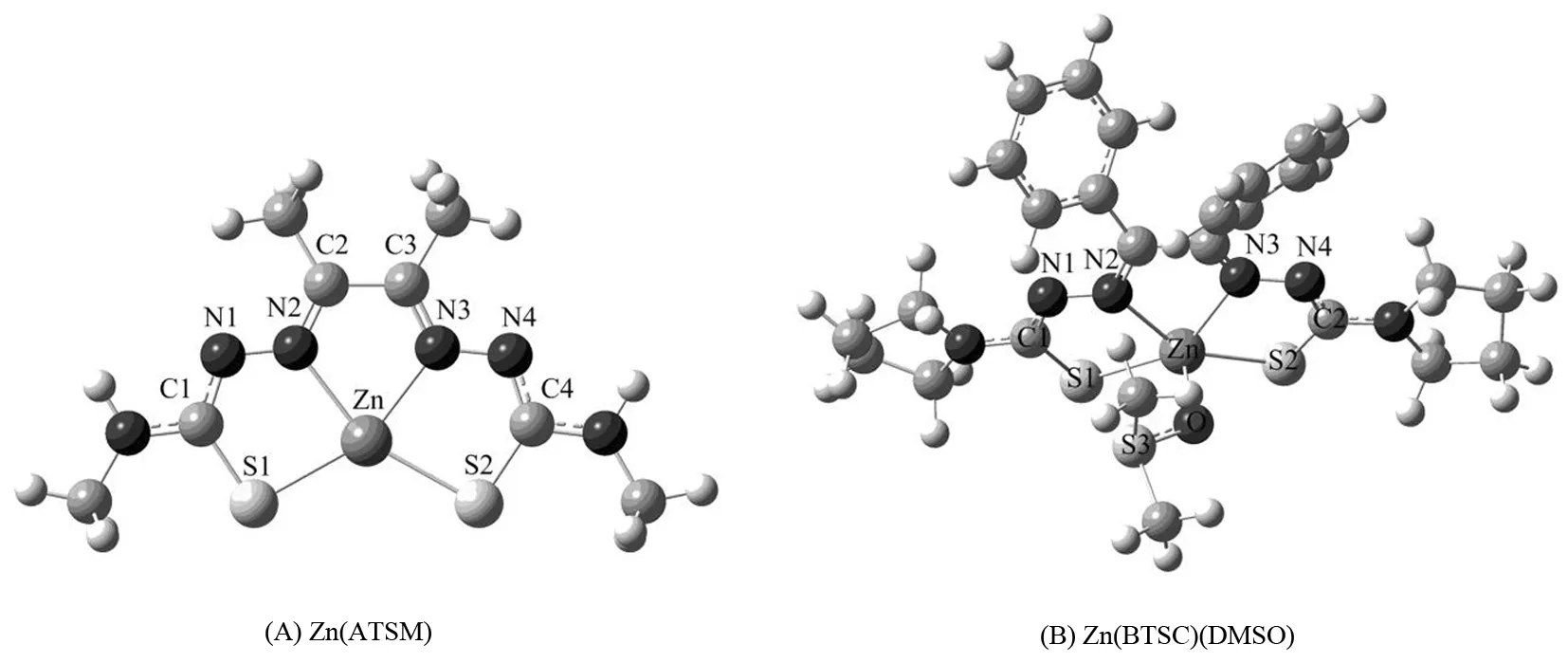
图1 配合物A和B的几何结构Fig.1 Optimized geometric structures of complexes A and B
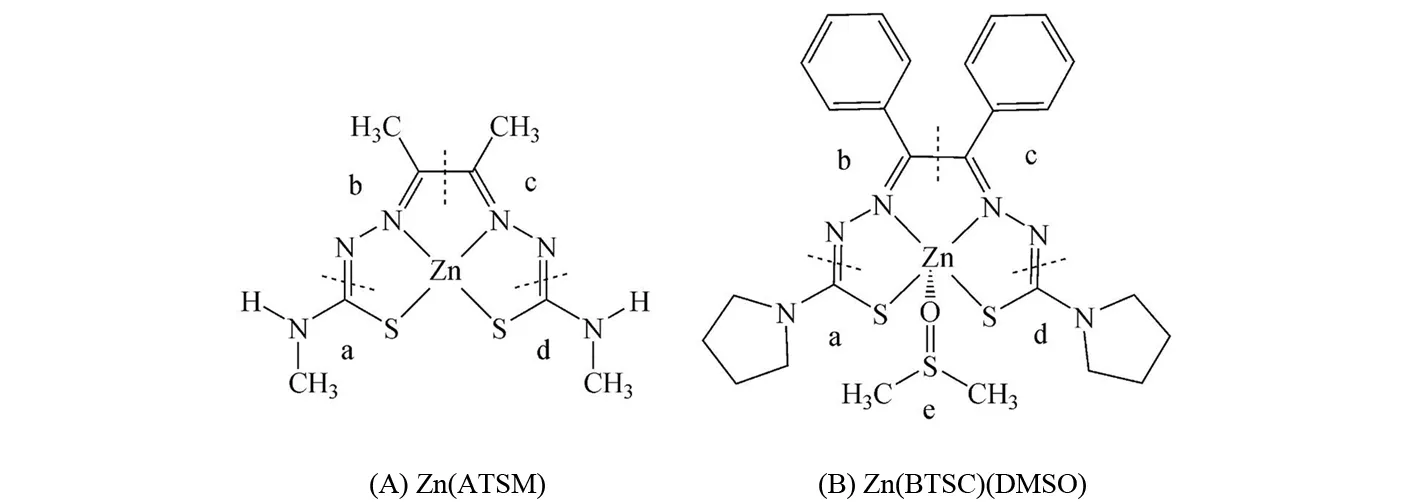
图2 配合物A和B的结构简图Fig.2 Schematic structures for complexes A and B
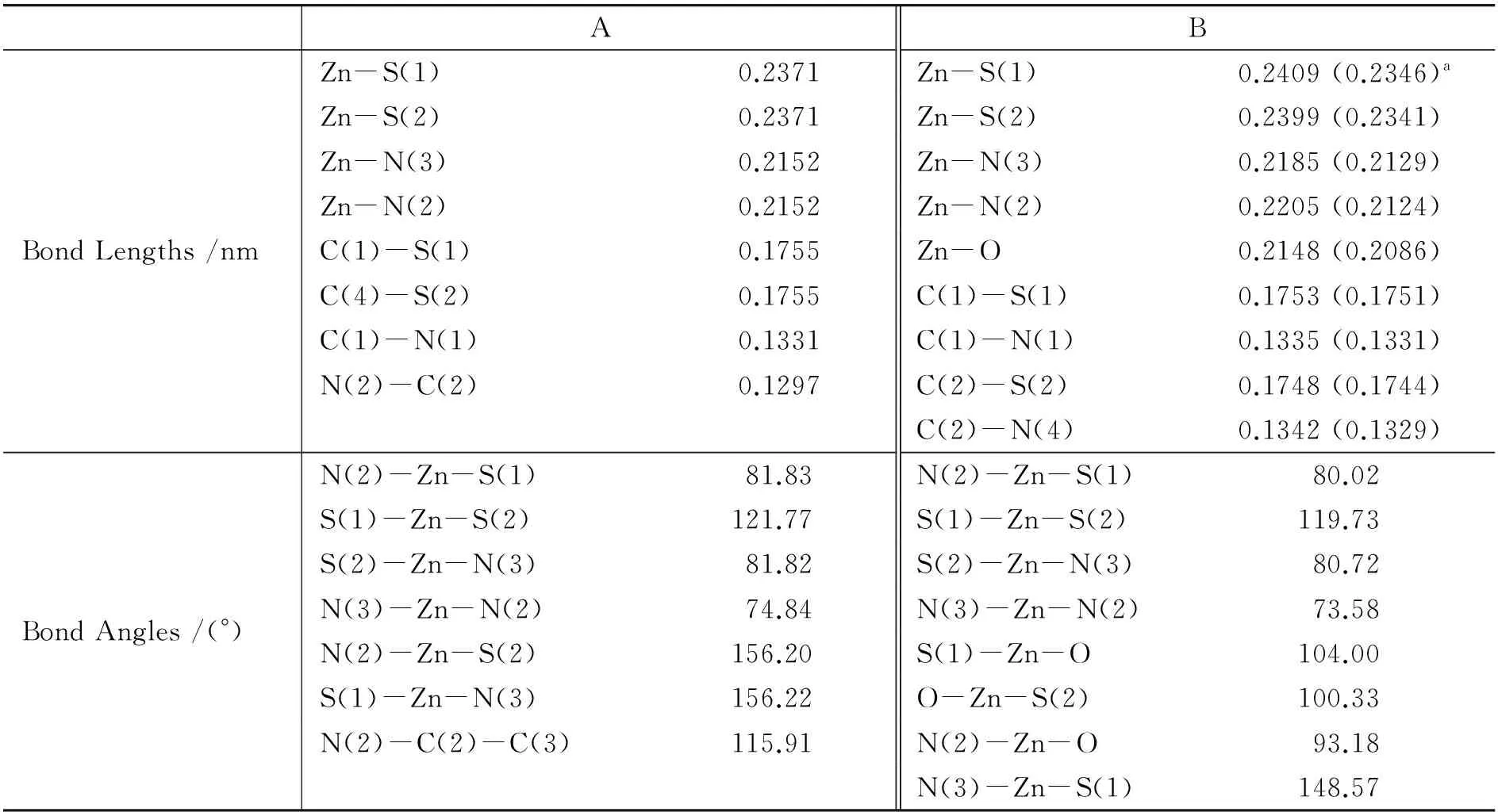
ABBondLengths/nmZn-S(1)0.2371Zn-S(1)0.2409(0.2346)aZn-S(2)0.2371Zn-S(2)0.2399(0.2341)Zn-N(3)0.2152Zn-N(3)0.2185(0.2129)Zn-N(2)0.2152Zn-N(2)0.2205(0.2124)C(1)-S(1)0.1755Zn-O0.2148(0.2086)C(4)-S(2)0.1755C(1)-S(1)0.1753(0.1751)C(1)-N(1)0.1331C(1)-N(1)0.1335(0.1331)N(2)-C(2)0.1297C(2)-S(2)0.1748(0.1744)C(2)-N(4)0.1342(0.1329)BondAngles/(°)N(2)-Zn-S(1)81.83N(2)-Zn-S(1)80.02S(1)-Zn-S(2)121.77S(1)-Zn-S(2)119.73S(2)-Zn-N(3)81.82S(2)-Zn-N(3)80.72N(3)-Zn-N(2)74.84N(3)-Zn-N(2)73.58N(2)-Zn-S(2)156.20S(1)-Zn-O104.00S(1)-Zn-N(3)156.22O-Zn-S(2)100.33N(2)-C(2)-C(3)115.91N(2)-Zn-O93.18N(3)-Zn-S(1)148.57
a括号中的数据为实验值[11].
2.2前线分子轨道
基态电子结构的变化对配合物的光化学性质有着重要的影响.因此,为了探究配合物A和B的激发态电子跃迁实质,我们分析了它们的最高占据轨道HOMO(H)和最低空轨道LUMO(L)的电子云分布情况.图3绘出了这两种配合物在吸收光谱中涉及到的部分前线分子轨道的能级和轮廓图.
根据配合物A和B的对称性,它们的结构可通过图2中的虚线分为a、b、c、d四部分.配合物A的HOMO能级为-5.61 eV,其电子云主要分布在c和b部分中氮原子的p轨道上.另外2个H-2和H-3占据轨道的电子云分别分布在a、b、c、d基团上和a、d基团上.配合物A的LUMO和L+9轨道上的电子云分布较为均匀,a、b、c、d基团对其都有一定的贡献.配合物B的占据轨道电子云分布特点与配合物A大致相同.然而,它的三个空轨道的电子云主要分布在b和c部分中苯基的π*轨道上.另外,与配合物A的HOMO和LUMO能级相比,配合物B的HOMO能级升高,LUMO能级降低.因此,配合物B的HOMO和LUMO之间的能级差减小,从而导致其最大吸收波长发生了红移.
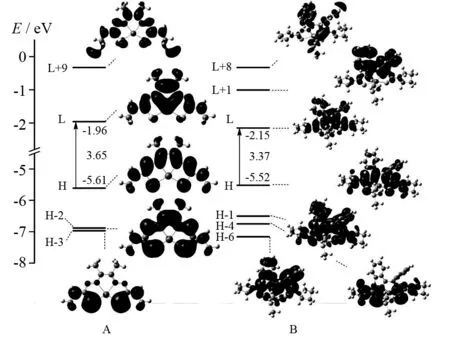
图3 配合物A和B的前线分子轨道组成能量以及H-L能级差Fig.3 Presentation of energy levels, energy gaps, and compositions of frontier molecular orbitals for complexes A and B
2.3吸收光谱
以优化的基态构型为基础,在TD-M06/6-31+G(d)-LANL2DZ水平下结合PCM模型计算了配合物A和B在二甲亚砜溶剂中的电子吸收光谱.表2中列出了配合物的垂直激发能(E)、吸收波长(λ)、振子强度(f)、主要电子组态、跃迁性质及相应的实验值.为了得到更直观的结果,图4模拟了配合物A和B的高斯型吸收曲线.
与配合物A相比,配合物B的最大吸收波长(482 nm)发生了红移.由于两者的S0→S1跃迁主要是由HOMO到LUMO的电子跃迁产生的,因此,该现象与它们的HOMO-LUMO能级差变化顺序是一致的.配合物A和B的最大吸收峰分别来自于Nb/Nc→Zn/Nb/Nc/Cb/Cc和Nb/Nc→Nb/Nc/Cb/Cc/Ph-c的跃迁,其中配合物A的H-L跃迁具有配体到金属的电荷转移(LMCT)/配体内电荷转移(ILCT)的特征,配合物B的H-L跃迁具有配体内电荷转移的特征(ILCT). 除此之外,配合物A和B在299 nm和344 nm处有一个较弱的吸收峰.配合物A在299 nm处的吸收峰主要来自于H-3→L的跃迁,配合物B在334 nm处的吸收峰主要是由H→L+1和H-6→L的跃迁产生的.由于配合物A的H-3轨道电子云主要分布在Sa/Na/Sd/Nd的p轨道上,而LUMO轨道上Nb/Cb/Nc/Cc的贡献较大,所以配合物A在299 nm处的跃迁为p→p跃迁,具有ILCT的转移特征.配合物B的H→L+1跃迁可归属为p→π*跃迁,而H-6→L的跃迁为(p, π)→(p, π*)的跃迁.配合物A在311 nm处还存在着一个振子强度较大的吸收峰(f=0.151 2).由此,我们推测实验上报道的配合物A在313 nm处的吸收峰可能是由299 nm和311 nm处的2个峰叠加形成的.同理,实验上配合物B在333 nm处的吸收可能是由359 nm和334 nm处的峰叠加产生的.另外,理论计算得到了两种配合物在实验上没有测得的吸收峰.例如,配合物B在277 nm处的吸收峰主要是由H→L+8的跃迁产生的.通过分析电子结构,发现配合物B的L+8轨道电子云主要分布在锌原子和Ca/Cd/Sd/Se/Ce/pyr-a/pyr-d的p轨道上及Ph-b/Ph-c的π*轨道上,而HOMO轨道上Nb/Nc的贡献较大.上述分析说明了配合物B的吸收峰是由ILCT、LLCT(配体到配体的电荷转移)和LMCT共同作用的结果.
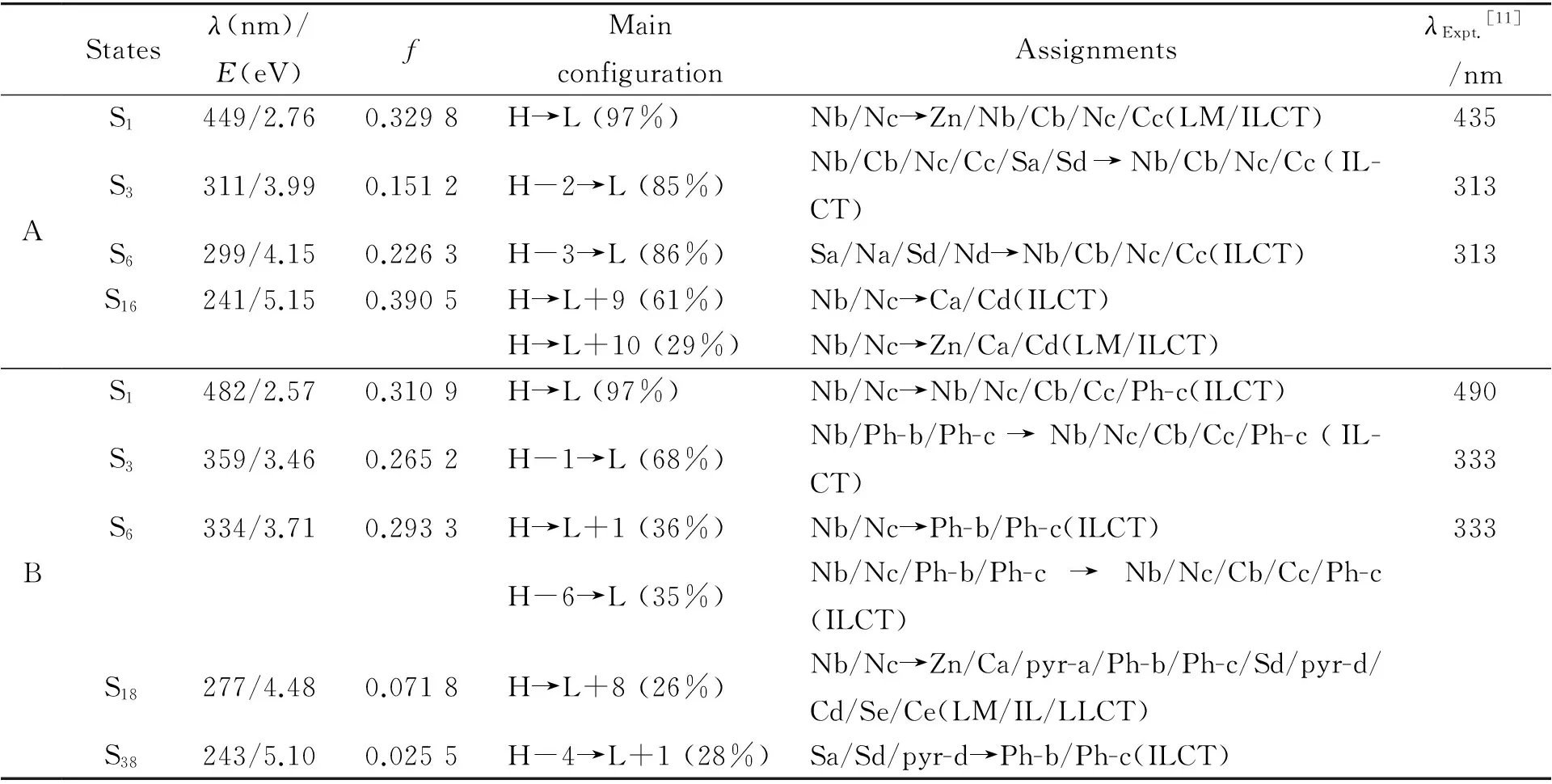
表2 在TD-M06方法下计算得到的配合物A和B在二甲亚砜溶剂中的吸收光谱数据及相应的实验值
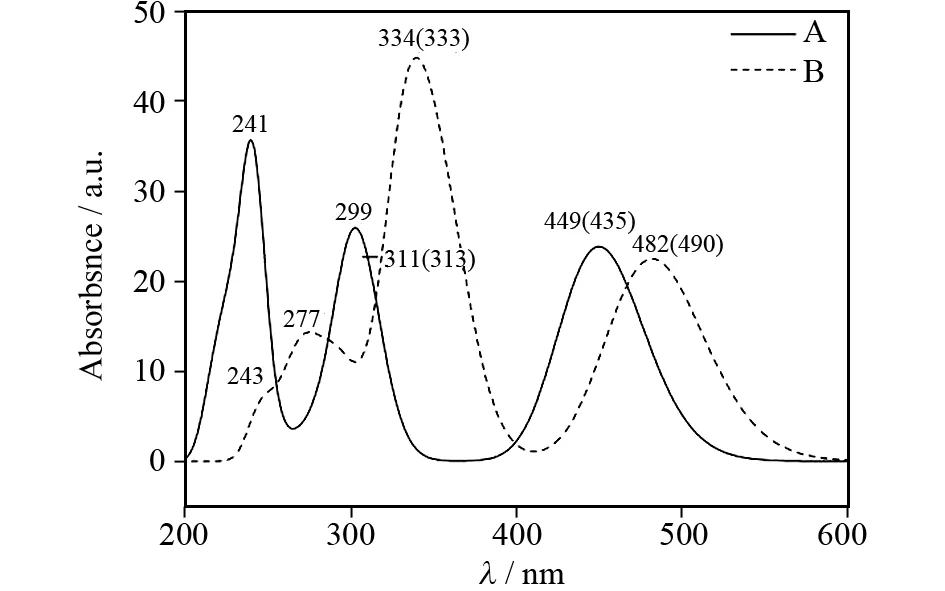
图4 在TD-M06 (PCM)水平下,配合物A 和B在二甲基亚砜溶剂中的吸收光谱曲线Fig.4 Simulated absorption spectra in DMSO medium for complexes A and B at the TD-M06 (PCM) level
3 结论
采用密度泛函理论(DFT)和含时密度泛函理论(TD-DFT),结合M06方法和6-31+G(d)-LANL2DZ基组研究了两种锌配合物A和B的几何构型和电子吸收光谱.计算结果表明,配合物B具有较小的HOMO-LUMO能级差,使其最大吸收波长相对于配合物A发生了红移.另外,采用含时密度泛函理论计算得到的电子吸收光谱与实验值吻合较好,验证了理论计算的可靠性,并得到了实验上没有测得的吸收峰.
[1] BLOUW B, SONG H, TIHAN T, et al. The hypoxic response of tumors is dependent on their microenvironment [J]. Cancer Cell, 2003, 4(2): 133-136.
[2] BROWN J M. The hypoxic cell: a target for selective cancer therapy-eighteenth Bruce F. Cain memorial award lecture [J]. Cancer Res, 1999, 59(23): 5863-5870.
[3] BROWN J M, WILSON W R. Exploiting tumour hypoxia in cancer treatment [J]. Nat Rev Cancer, 2004, 4(6): 437-447.
[4] TATUM J L, KELLOFF G J, GILLIES R J, et al. Hypoxia: importance in tumor biology, noninvasive measurement by imaging, and value of its measurement in the management of cancer therapy [J]. Int J Radiat Biol, 2006, 82(10): 699-757.
[5] ISA A Y, WARD T H, WEST C M, et al. Hypoxia in head and neck cancer [J]. Brit J Radiol, 2006, 79(946): 791-798.
[6] CARROLL L, BEJOT R, HUETING R, et al. Orthogonal18F and64Cu labelling of functionalised bis(thiosemicarbazonato) complexes [J]. Chem Commun, 2010, 46(23): 4052-4054.
[7] FERREIRA M F, MARTINS A F, MARTINS J A, et al. Gd(DO3A-N-alpha-aminopropionate): a versatile and easily available synthon with optimized water exchange for the synthesis of high relaxivity, targeted MRI contrast agents [J]. Chem Commun, 2009(42): 6475-6477.
[8] HOLLAND J P, FISHER V, HICKIN J A, et al. Pyrene-functionalised copper complexes as potential dual-modality imaging agents [J]. Eur J Inorg Chem, 2010, 2010(1): 48-58.
[9] COWLEY A R, DAVIS J, DILWORTH J R, et al. Fluorescence studies of the intra-cellular distribution of zinc bis(thiosemicarbazone) complexes in human cancer cells [J]. Chem Commun, 2005(7): 845-847.
[10] CHRISTLIEB M, HOLLAND J P, DILWORTH J R. Investigation of the UV-vis absorption of bis(N-methylthiosemicarbazonato) zinc Zn [ATSM] [J]. Inorg Chim Acta, 2010, 363(6): 1133-1139.
[11] DAYAL D, PALANIMUTHU D, SHINDE S V, et al. A novel zinc bis (thiosemicarbazone) complex for live cell imaging [J]. J Biol Inorg Chem, 2011, 16(4): 621-632.
[12] ZHAO Y, TRUHLAR D G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals [J]. Theor Chem Acc, 2008, 120(1/3): 215-241.
[13] HEHRE W J, DITCHFIELD R, POPLE J A. Self-consistent molecular orbital methods. XII. Further extensions of Gaussian-type basis sets for use in molecular orbital studies of organic molecules [J]. J Chem Phys, 1972, 56(5): 2257-2261.
[14] WADT W R, HAY P J. Ab initio effective core potentials for molecular calculations. Potentials for main group elements Na to Bi [J]. J Chem Phys, 1985, 82(1): 284-298.
[15] BAUERNSCHMITT R, AHLRICHS R. Treatment of electronic excitations within the adiabatic approximation of time dependent density functional theory [J]. Chem Phys Lett, 1996, 256(4): 454-464.
[17] TOMASI J, PERSICO M. Molecular interactions in solution: an overview of methods based on continuous distributions of the solvent [J]. Chem Rev, 1994, 94(7): 2027-2094.
[18] FRISCH M J, TRUCKS G W, SCHLEGEL H B, et al. Gaussian 09 [CP]. Revision A. 02, Wallingford CT: Gaussian, Inc., 2009.
[责任编辑:吴文鹏]
Theoretical investigation of the electronic structure and spectral properties of two zinc (Ⅱ) complexes
WANG Xin, LI Yonghong*
(InstituteofEnvironmentalandAnalyticalSciences,CollegeofChemistryandChemicalEngineering,HenanUniversity,Kaifeng475004,Henan,China)
To elucidate the electronic structure information and spectral properties, the ground-state structures of Zn(Ⅱ) complexes Zn(ATSM) (A) and Zn(BTSC)(DMSO) (B) were optimized by M06 method. The calculated electronic structure was used to generate a qualitative picture of the changes in electron distribution, which occurs during the absorption process. The theoretical absorptions agree well with the experimental results. Moreover, some distinguishable absorption peaks that were not reported in experiment are detected in theory.
DFT; absorption spectrum; Zn(Ⅱ) complexes
1008-1011(2016)04-0445-05
2016-05-17.
河南省教育厅自然科学研究项目(14A150034).
汪欣(1992-), 男, 硕士生, 研究方向为量子化学计算.*通讯联系人,E-mail: liyonghong@henu.edu.cn.
O641.3
A
