拉曼光谱在天然纤维素结构研究中的应用进展
2016-07-12马建锋杨淑敏田根林刘杏娥
马建锋,杨淑敏,田根林,刘杏娥
国际竹藤中心,北京 100102
拉曼光谱在天然纤维素结构研究中的应用进展
马建锋,杨淑敏,田根林,刘杏娥*
国际竹藤中心,北京 100102
纤维素是木质纤维生物质细胞壁的骨架物质,也是生物燃料制备过程中重要的前驱体。作为重要的天然有机高分子,纤维素分子结构的研究备受关注。拉曼光谱仪因其较高的分辨率及无损检测的特点可在多尺度研究天然纤维素复杂分子链及聚集态结构。本文在比较了色散型拉曼光谱仪和傅里叶变换拉曼光谱仪的构造及相关参数的基础上,详细综述了拉曼光谱技术在植物细胞壁纤维素微区分布、天然纤维素酶解发酵、分子链空间取向、分子形变、结晶度与多晶态转变等方面的研究进展。并对拉曼光谱技术在天然纤维素分子结构研究中存在的问题进行了总结,提出了可能的解决方案,以促进拉曼光谱技术在天然有机高分子研究领域的应用。
天然纤维素; 拉曼光谱; 高分子链结构; 聚集态结构
引 言
随着化石资源的消耗殆尽和环境保护意识的增强,可再生资源的开发和应用已成为国内外竞相开展的研究课题。作为棉花、木材、亚麻、草类等高等植物细胞壁的主要成分,纤维素主要由植物通过光合作用合成,是自然界取之不尽,用之不竭的可再生资源。研究表明纤维素是由D-葡萄糖以β-1,4糖苷键联接而成的链状高分子化合物,同时纤维素分子内和分子间存在着大量的氢键,形成复杂的高分子链结构和聚集态结构。复杂的高分子链结构直接影响着纤维素及纤维素复合材料的物理(润胀、吸湿、热塑性)以及力学(拉伸强度、弹性模量)性能,纤维素超分子结构中存在的结晶区及非晶区两相聚集态结构直接影响纤维素的化学反应性能。当前,纤维素化工行业存在的能耗高、试剂用量大、产物性能不稳定等问题,均与纤维素分子链和聚集态超分子结构的性质密切相关。另外,在生物质转化为生物乙醇的过程中,需使用纤维素酶先将纤维素降解为单糖,而后将单糖发酵制备生物乙醇。在线监控纤维素分子水解生成葡萄糖的过程,可以指导纤维素酶用量,进而降低生物乙醇生产的成本。综上所述,纤维素分子结构的研究对天然纤维原料的开发利用具有重要的意义。
传统研究纤维素高分子结构的方法主要有X射线衍射(XRD)、中子散射以及交叉极化魔角旋转核磁共振波谱(CP-NMR)技术。这些方法在研究纤维素分子结构时,通常需要对纤维素样品进行物理或是化学预处理,处理过程不可避免地会改变纤维素样品天然状态下的分子结构。相比较而言,分子光谱能够有效地在原位状态下研究纤维素样品的分子结构。早在1947年红外光谱就被用于纤维素分子结构的研究,但样品中水分的存在极大地限制了红外光谱在纤维素结构研究中的应用。另外,由于衍射极限的限制显微红外光谱仅能提供组织水平的分子结构信息。而作为分子光谱中的另一个重要分支,拉曼光谱因其制样简易及较高的空间和光谱分辨率在纤维素分子结构研究中得到了广泛的应用。本文比较了两种主要类型拉曼光谱仪的构造特点,同时详细综述了拉曼光谱仪在天然纤维素研究领域的应用进展,以期为纤维素生物合成、纤维素高效分离以及纤维素基新产品的开发提供新的研究思路和手段。
1 拉曼光谱仪
激光拉曼光谱仪按分光系统差异主要分为两种: 色散型拉曼(dispersive Raman, DIS-Raman)和傅里叶变换拉曼(fourier transform Raman, FT-Raman)。色散型拉曼光谱仪主要通过单极或是多级光栅进行分光,而傅里叶变换拉曼采用迈克尔逊干涉仪进行分光。
色散型激光拉曼光谱仪主要由以下几个部分组成(如图1): 激光光源→样品室→色散系统→检测器→数据处理系统。

图1 色散型拉曼光谱仪基本结构
傅里叶变换拉曼光谱仪与色散型拉曼光谱仪不同,它主要由以下几个部分组成(图2): 激光光源→样品室→相干滤波器→干涉仪→检测器→计算机处理数据(进行傅里叶变换)。

图2 傅里叶变换型拉曼光谱仪基本结构
由于构造原理的差异,色散型拉曼和傅里叶变换拉曼的性能有所不同,两者的比较如表1所示。色散型拉曼通常采用532 nm(可见光)、633 nm(可见光)和785 nm(近红外)半导体激光器,特别是785 nm,与可见光及紫外激光器相比,能极大地克服荧光问题。同傅里叶变换拉曼相比,色散型拉曼采用的CCD检测器具有更高的量子效率,因此具有更高的灵敏度和检测限。1990年,色散型拉曼光谱仪首次与显微镜结合并被命名为显微拉曼光谱(Micro-Raman-spectroscopy)。它将激发光通过显微镜聚焦在直径为数微米的光斑上,在这一微小区域内获得拉曼光谱,利用显微镜具有较高的数值孔径(NA),显微拉曼光谱能够极大地提高拉曼信号的采集效率。但采用该法检测样品时,由于激光被聚焦在数微米的光斑内,可能会导致样品因受热而破坏。
绝大部分的傅里叶变换拉曼光谱仪都采用1064 nm半导体激光器作为激发光,以减少激光诱导产生的荧光信号。由于原理上的优势,傅里叶变换拉曼光谱仪具有较高的光谱分辨率,更易于和傅里叶变换拉红外光谱仪联用,适合于对光谱分辨率有较高要求的科学研究中。
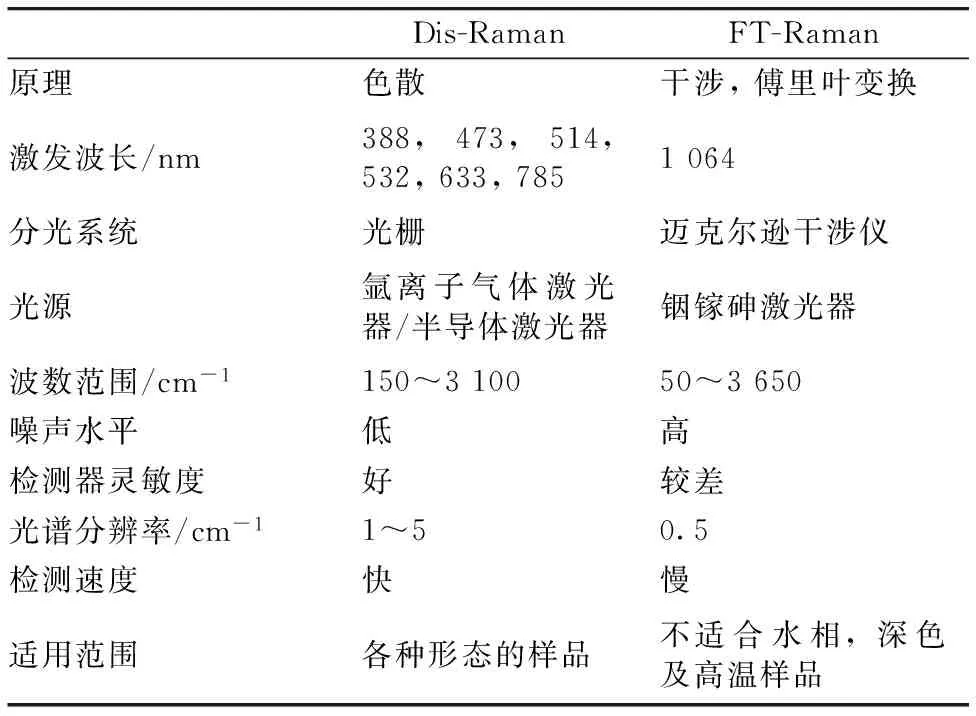
表1 色散型及傅里叶变换型拉曼光谱仪比较
2 天然纤维素拉曼光谱特征峰归属
拉曼光谱仪应用于天然纤维素的研究首先是要对纤维素拉曼光谱中各个特征峰进行归属,通过拉曼光谱峰位的归属分析可以得到官能团、化学键和电子密度等分子结构及其变化信息。拉曼光谱峰位归属结果列于表2中。其中2 895 cm-1特征峰归属于纤维素CH2伸缩振动,而在植物细胞壁中这一特征峰通常包含了半纤维素的贡献,因此,用来反映细胞壁中碳水化合物的浓度空间差异。1 096 cm-1特征峰归属于纤维素糖苷键(C—O—C)不对称伸缩振动,1 122 cm-1特征峰归属于纤维素糖苷键(C—O—C)对称伸缩振动,其中1 098 cm-1特征峰的强度受拉曼光谱仪入射光偏振方向影响极为明显,因此,这两个特征峰的比值反映了天然纤维素的空间取向差异。

表2 天然纤维素拉曼光谱特征峰归属
3 天然纤维素拉曼光谱研究
3.1 天然纤维素分子拉曼光谱研究
3.1.1 纤维素微区分布研究
拉曼光谱研究天然纤维素微区分布,是通过对拉曼光谱中纤维素分子特征峰的峰高、峰宽或峰面积积分获得光谱成像图实现的。通过分析获得的拉曼光谱成像图,可以定性地研究纤维素浓度的空间差异。在植物细胞壁组分分布研究中,纤维素空间分布图像可以通过对拉曼光谱中345~390 cm-1(β-D-葡萄糖环)、978~1 178 cm-1(C—O—C)和2 810~2 936 cm-1(C—H,C—H2)三个波数区域积分获得。针叶木管胞、阔叶木纤维细胞以及禾本科厚壁纤维细胞的拉曼光谱成像图表明纤维素主要沉积在细胞的次生壁中,且浓度沿相邻的细胞次生壁成波动规律,在胞间层区域浓度最低[1-3]。然而,杨木受拉木(由于强风和地心引力作用,形成于阔叶木倾斜枝干上端的应变组织)中的纤维素微区分布较为特殊,研究发现受拉木中纤维细胞凝胶层的纤维素浓度高于邻近的次生壁及胞间层。由于2 810~2 936 cm-1含有半纤维素的信号,978~1 178 cm-1波数区域包含的1 096 cm-1特征峰受入射光偏振方向影响,因此在纤维素微区分布研究中采用345~390 cm-1波数区域较为合理,尽管这一区域的信号强度较弱。
3.1.2 纤维素分子酶水解研究
在木质纤维原料中,细胞壁中的纤维素经酶水解可以转化为单糖,这些单糖进一步发酵可以制成生物乙醇。目前分析水解液中单糖的主要方法有高效液相色谱,紫外可见光谱,气相色谱-质谱联用以及电化学方法。相比以上分析方法,拉曼光谱能够在无损条件下研究水解液中各种类型单糖浓度。Shih和Smith[4]利用拉曼光谱仪定量地研究了生物炼制过程中纤维素转化为单糖以及单糖发酵制备生物乙醇的效率。其中葡萄糖和乙醇定量研究时选用的特征峰分别为1 090cm-1(C—C伸缩振动)和883 cm-1(C—C伸缩振动),两者的拉曼谱图信号检测限分别为8和6 g·L-1,同时比较不同生物质预处理方法对纤维素酶水解效率影响时发现,只有能够去除木质素的预处理方法才能利用拉曼光谱技术计算转化效率。Shih等[5]进一步结合化学计量学中的去卷积和多峰拟合光谱处理方法定量的研究了生物质酶水解过程中葡萄糖(积分区域519 cm-1)和木糖(积分区域536 cm-1)的浓度。该方法在总糖浓度高于45 mg·mL-1时的标准差为6.1%,同时发现水、乙醇、己烷抽提能够改善拉曼光谱背景,有助于单糖检测限提高。尽管拉曼光谱已经成功的应用于监测生物质预处理及酶解发酵过程中单糖浓度变化,但是诸多因素仍然限制了其更广泛的应用,这其中包括,预处理液体中抑制物产生的荧光背景,各种小分子对葡萄糖、木糖、乙醇拉曼信号的干扰。因此,进一步推广这一技术在生物质酶解发酵研究领域的应用,依赖于对预处理及水解液中荧光背景来源的详细研究以及排除小分子对单糖信号的干扰。
3.2 天然纤维素高分子链拉曼光谱研究
3.2.1 纤维素分子链空间取向
纤维素分子是由D-葡萄糖以β-1,4糖苷键联接而成的链状高分子化合物。纤维素分子链在氢键和范德华力作用下平行排列形成原细纤维,若干根原细纤维组成微细纤维,微细纤维连同周围的半纤维素和木质素一起,组成了细胞壁的细纤维。通过研究细胞壁中纤维素的高分子链取向,可以近似反映微细纤维(微纤丝)的空间排列方式。植物细胞壁中联接纤维素吡喃环的糖苷键(C—O—C)为平伏键,而次甲基(C—H)为直立键。这两个键对拉曼光谱入射光偏振方向极其敏感,它们的强度与激光的偏振方向存在明显的相关性。因此利用增加偏振片附件的显微拉曼光谱仪可以在细胞甚至亚细胞水平研究纤维素高分子链/微纤丝的空间取向。Atalla等[6]利用偏振光拉曼光谱技术研究了苎麻(纤维素高分子链均平行于细胞轴)中纤维素的糖苷键(C—O—C,1 098 cm-1)和次甲基(C—H, 2 920 cm-1)强度与入射光方向变化的关系,结果表明,当入射光电矢量方向平行细胞轴向时,1 098 cm-1特征峰较强,而垂直于细胞轴时这一特征峰强度明显降低。2 920 cm-1特征峰强度变化规律与此相反。随后Agarwal和Atalla[7]利用偏振光显微拉曼光谱技术研究了云杉管胞次生壁中纤维素高分子链的空间取向,在管胞弦切面上分别以平行和垂直于细胞轴的偏振光电矢量方向进行信号采集,结果发现,当入射光电矢量方向趋近平行于细胞轴方向变化时,C—O—C特征峰相对强度增加,而此时C—H,C—H2特征峰相对强度降低。由此可知云杉管胞纤维素高分子链中C—O—C趋近平行于细胞轴排列方向。以上研究表明,当拉曼光谱仪激光电矢量偏振方向平行于纤维素高分子链时,糖苷键(1 098 cm-1)特征峰强度增加,而趋近垂直于高分子链时强度逐渐降低。
基于以上结果,Ma等[8]系统地报道了灌木红瑞木杂细胞(导管、薄壁细胞)与纤维细胞的微纤丝空间取向差异。结果表明,相对于纤维细胞次生壁,导管细胞次生壁中纤维素微纤丝排列更趋近垂直于细胞轴(图3)。然而,以上研究均是定性地阐述纤维素微纤丝的空间分布,为了突破定量研究微纤丝空间取向这一技术瓶颈,Gierlinger等[9]结合二次线性回归和偏最小二乘法定量地计算了云杉对应木和应压木管胞次生壁外层S1和中层S2的微纤丝角,且此计算结果与X射线衍射法测量结果相近似。虽然显微拉曼光谱技术能够定量地研究植物细胞壁中纤维素微纤丝的空间取向,但这一技术的广泛应用受到样品自身特性的限制,利用这一技术首先要选择微纤丝角小于10°且厚度大于激光光斑(0.5~1.0 μm)的细胞次生壁中层建立模型。然而在植物界中同时满足这两种要求的细胞种类少之又少。鉴于植物细胞壁复杂的壁层结构,准确测定不同壁层的微纤丝角方向仍是植物细胞生物学未来研究的重点之一。

图3 红瑞木纤维及导管细胞中纤维素 微纤丝空间取向拉曼光谱成像图
Fig.3 Raman spectroscopic imaging showing the orientation of cellulose microfibrils in the fiber and vessel ofCornusalba, 1 091~1 100 cm-1
3.2.2 纤维素高分子链形变
拉曼光谱主要反映的是分子化学键振动类型及能量的差异,当分子受到外力作用时必将导致化学键的形变,而形变的程度可以通过拉曼光谱特征峰峰位偏移进行定量的描述。Gierlinger等[10]利用拉曼光谱研究了单根纤维拉伸过程中纤维素糖苷键(C—O—C)拉曼特征峰1 097 cm-1偏移与拉伸应力间的关系。结果表明拉伸应力与1 097 cm-1位移存在明显的相关性(r=0.99),拉伸过程中1 097 cm-1的拉曼偏移量为-6.5 cm-1,偏移的速率为-6.1 cm-1/GPa。而当单根纤维拉断后纤维素糖苷键拉曼特征峰的位移又恢复到1 097 cm-1,表明了单根纤维的弹性特质。另外,在拉伸过程中O—H区域(3 375~3 402 cm-1)也发生了明显的拉曼偏移,这主要归因于纤维素分子中氢键网络的破环。因此,利用1 097 cm-1特征峰偏移量以及特征峰的强度比可以用于研究天然纤维及纤维素复合材料的形变量。在此基础上Ma等[11]在细胞水平研究了黑杨受拉木纤维细胞不同细胞壁层的形变特征,结果表明次生壁及凝胶层拉曼光谱中1 097 cm-1拉曼特征峰向低波数偏移了3 cm-1,这一结果从分子水平解释了受拉木拉伸应力产生的原因。而与单根纤维拉伸试验不同的是Ma等的研究结果表明受拉木凝胶细胞壁中的纤维素糖苷键在拉伸过程中产生了永久的分子形变。尽管拉曼光谱已经成功地应用于单根纤维及细胞壁应力变化研究,然而实时观测单根纤维拉伸过程中应力变化时,纤维形变不可避免地会造成显微拉曼光谱仪焦点的变化,进而影响拉曼光谱中特征峰的信号强度。因此利用拉曼光谱研究单根纤维拉伸形变时,若试图利用特征峰强度解释应力变化,应将焦点改变造成的误差考虑在内。
3.3 天然纤维素聚集态结构研究
3.3.1 纤维素结晶度
纤维素结晶度对生物质材料的物理、力学以及化学性质具有明显的影响。随着结晶度的增加,生物质材料的拉伸应力、尺寸稳定性以及密度增加,而化学反应活性及润胀程度降低。另外,在生物质炼制过程中较高的纤维素结晶度对酶水解具有抑制作用。因此快速、简便、准确地测定纤维素结晶度对木质纤维原料的利用尤为重要。纤维素大分子由结晶区和非结晶区交替组成,通过混合不同比例的结晶纤维素Ⅰ及无定形纤维素,利用拉曼特征峰的相对强度可以表征混合物的结晶度。比较高结晶度的纤维素Ⅰ(冷冻干燥后的细菌纤维素)和无定形纤维素的拉曼光谱得知结晶和无定形态的纤维素I的拉曼光谱差异主要出现在对构像敏感的低频区(1 500~950 cm-1),该区域存在纤维素骨架吡喃环C—C,C—O,C—O—C伸缩振动以及HC—C,HC—O,C—OH和C—H2弯曲振动特征峰。比较发现结晶和无定形纤维素Ⅰ拉曼光谱可以发现两者分别在1 481和1 462 cm-1出现特征峰 基于这一差异,Schenzel等[12]利用拉曼光谱中纤维素Ⅰ的亚甲基弯曲振动特征峰1 481 cm-1(结晶纤维素)和1 462 cm-1(无定型纤维素)强度比值建立了纤维素Ⅰ的拉曼结晶指数(XCRaman=I1 481 cm-1/I1 462 cm-1+I1 481 cm-1)。但是纤维素Ⅰ拉曼光谱中1 481和1 462 cm-1特征峰强度较低,且光谱拟合过程中会引入误差。因此,Umesh等[13]利用单变量及多变量分析法建立了新的纤维素I拉曼结晶指数(XCRaman=[(I380 cm-1/I1 096 cm-1)-0.028 6]/0.006 5)。在进一步研究针叶木、阔叶木、纸浆及各种农林残余物在内的41种天然纤维的拉曼结晶指数时,Umesh等[14]发现紫丁香基木质素在370 cm-1处存在的特征峰会使测得的结晶指数高于真实值,而半纤维在1 096 cm-1处存在的特征峰会使测得的拉曼结晶指数偏低。在研究纤维测试位置及样品尺寸对结晶度影响的实验中,Umesh等[15]发现短叶松木块相对于激光的电矢量方向变化及木粉颗粒大小对结晶度的影响较小。尽管拉曼光谱已经逐步应用于纤维素结晶度的计算,但是将其直接应用于木质纤维原料(木材、竹材以及草类等)还有待进一步研究,研究的重点是探讨如何去除半纤维素、木质素以及荧光背景对光谱中纤维素结晶度相关特征峰的干扰。
3.3.2 纤维素多晶态转变
纤维素是一种同质多晶体,现在已知的纤维素结晶变体有纤维素Ⅰ(天然纤维),Ⅱ(丝光化纤维素或是再生纤维素),Ⅲ,Ⅳ以及ⅢⅠ,ⅢⅡ,ⅣⅠ,ⅣⅡ。不同的晶型结构对纤维素的物理化学性质有重要的影响,因而被广泛研究。早在1975年Atalla和Dimick[16]就利用拉曼光谱研究了纤维素Ⅰ、纤维素Ⅱ和纤维素Ⅲ1的分子结构差异。比较三者的拉曼光谱可以发现,纤维素Ⅱ和纤维素ⅢⅠ的拉曼光谱近似,而与纤维素Ⅰ存在明显差异。在O—H伸缩振动区域α-纤维素Ⅱ和α-纤维素ⅢⅠ分别在3 490和3 482 cm-1波数区出现拉曼特征峰,而α-纤维素Ⅰ在这一区域没有特征峰出现,这很可能是由于分子间或分子内的氢键差异造成的。在C—H伸缩振动区域三者的拉曼特征峰位置各不相同,这反映出三种纤维素的空间构型差异。在C—H剪切振动区域(1 400~1 500 cm-1),纤维素Ⅰ在1 476和1 461 cm-1出现两个特征峰,而纤维素Ⅱ和纤维素ⅢⅠ仅在1 461 cm-1出现一个特征峰。其中1 476和1 461 cm-1分别归属于纤维素碳六位置上CH2OH基团的tg和gg构型。这表明纤维素Ⅰ具有tg和gg构型,而纤维素Ⅱ和纤维素Ⅲ1仅有gg构型。在C—H剪切振动的其他区域,维素Ⅱ和纤维素Ⅲ1分别在1 265,697,577,420 cm-1出现拉曼特征峰,这些特征峰在纤维素Ⅰ中没有出现。因此利用这些特征峰位置的差异可以快速无损地区分纤维素Ⅰ与纤维素Ⅱ。比较纤维素Ⅱ和纤维素Ⅲ1拉曼光谱可以发现纤维素Ⅲ1分别在3 352,2 922,1 420,1 048,550,436,105 cm-1出现拉曼特征峰,而纤维素Ⅱ中拉曼特征峰出现在3 446,3 329,2 984,2 956,2 935,2 911,2 744,1 337,491 cm-1。
在纤维素转化利用过程中,构象的转变具有重要意义。Schenzel等[17]以不同比例混合纤维素Ⅰ和纤维素Ⅱ,利用拉曼光谱研究了纤维素多晶态的转变过程。结果发现随着纤维素Ⅱ比例的增加,纤维素Ⅰ中1 477和1 455 cm-1拉曼特征峰逐渐消失,在纯的纤维素Ⅱ样品中,仅仅1 461 cm-1处出现拉曼特征峰。当纤维素Ⅰ向纤维素Ⅱ的晶格转变过程中CH2OH基团的扭转振动方式也会发生变化,这导致了纤维素Ⅰ1 294 cm-1转变成纤维素Ⅱ中的1 465 cm-1拉曼特征峰。在转化过程中葡萄糖环振动模式也发生了变化。具体表现在纤维素Ⅰ和纤维素Ⅱ拉曼光谱中379和352 cm-1拉曼特征峰相对强度的变化。
在丝光化(所谓“丝光化”是指用一定浓度的碱液处理纤维素纤维的过程。纤维经丝光化处理后将发生不同程度的物理和化学变化,在碱的作用下,纤维得到充分润胀,碱液扩散至纤维细胞壁内部,使纤维素结晶区间的半纤维素、树脂和色素等都能较好地溶解而除去)过程中纤维素也会发生多晶态的转变,即由纤维素Ⅰ转变为纤维素Ⅱ,且这一过程是不可逆的。Jähn等[18]研究麻纤维丝光化过程中,分析拉曼光谱中亚甲基弯曲振动区域(1 400~1 475 cm-1)发现当碱溶液浓度超过15%时纤维素Ⅰ全部转变为纤维素Ⅱ,在拉曼光谱中仅1 460 cm-1处出现拉曼特征峰。
Schenzel等[19]利用傅里叶变换拉曼光谱研究了不同浓度碱(0%~16%)溶液处理纤维Ⅰ过程中纤维素的多晶态转化过程,结果表明丝光化处理过程中纤维素Ⅰ2 893 cm-1拉曼特征峰向低波数方向偏移了13 cm-1,且当碱溶液浓度超过12%时开始发生纤维素Ⅰ向纤维素Ⅱ的转变。将纤维素Ⅰ溶解在无机盐的水溶液(ZnCl2·4H2O,LiSCN·2,5 H2O,LiCl·ZnCl2·6H2O),通过拉曼光谱也可以观察到纤维素Ⅰ向纤维素Ⅱ的多晶态转变过程。溶解后的纤维素糖苷键拉曼特征峰向低波数方向偏移10 cm-1。且溶解后的纤维素拉曼光谱与无定形纤维素拉曼光谱类似。
4 展 望
拉曼光谱已经广泛应用于天然纤维素结构研究领域的各个方面,通过纤维素拉曼光谱不仅可以有效观测植物细胞壁内纤维素的微区分布、拉伸状态,还可以实时检测纤维素酶水解过程中葡萄糖和木糖的浓度。在丝光化过程中纤维素内化学键的变化和分子间相互作用也能通过拉曼光谱进行阐释。而这些研究大多数是基于定性的分析,而定量研究的相关报道还很少。在今后的研究中,研究人员应充分利用化学计量学的方法如偏最小二乘分析,主成分分析等方法定量或半定量地获取拉曼光谱背后隐藏的信息。除此之外,大部分色散型拉曼系统采用背散射采集方式获取拉曼信号,一般只能获得样品表面的信息,而透射拉曼可获得样品三维体积内的整体信息。因此,可以定量研究纤维素浓度,同时满足在同一台仪器上实现显微测试和整体测试的目的。在纤维素空间分布研究中,共聚焦显微拉曼光谱的空间分辨率是主要限制因素,当与原子力显微镜联用后构成的针尖增强拉曼光谱仪(Tip Enhanced Raman Spectroscopy)能够突破衍射极限将空间分辨率提升至纳米水平,这为在纳米尺度研究生物质细胞壁的化学信息提供了可能。目前关于纤维素分布的研究主要集中在二维平面空间,而纤维素的分布在三维空间是存在差异的,对纤维素在三维空间微区分布及溶解规律的探索必将是未来植物显微领域研究的前沿科学问题。在纤维素聚集态研究过程中,由于氢键体系为较弱的分子间相互作用,因此探索超低波数区域(10~20 cm-1),有望观测到天然以及化学预处理过程中纤维素构象、晶型、结晶度的精细变化信息。由此,我们可以看出拉曼光谱仪必将在生物质转化、植物细胞生物学以及材料科学研究领域发挥越来越重要的作用。
[1] Agarwal U P. Planta, 2006, 224(5): 1141.
[2] Richter S, Mussig J, Gierlinger N. Planta, 2011, 233(4): 763.
[3] Gierlinger N, Sapei L, Paris O. Planta, 2008, 227(5): 969.
[4] Shih C Ju, Smith E A. Analytica Chimica Acta, 2009, 653(2): 200.
[5] Shih C Ju, Lupoi J S, Smith E A. Bioresource Technology, 2011, 102(8): 5169.
[6] Atalla R H, Rebecca E, Whitmore C. Macromolucules, 1980, 13(6): 1717.
[7] Agarwal U P, Atalla R H. Planta, 1986, 169(3): 325.
[8] Ma J F, Ji Z, Zhou X, et al. Microscopy and Microanalysis, 2013, 19(1): 243.
[9] Gierlinger N, Luss S, Konig C, et al. Journal of Experimental Botany, 2010, 61(2): 587.
[10] Gierlinger N, Schwanninger M, Reinecke A, et al. Biomacromolecules, 2006, 7(7): 2077.
[11] Ma J F, Zhou X, Zhang X, et al. Bioresources, 2013, 8(2): 2222.
[12] Schenzel K, Fischer S, Brendler E. Cellulose, 2005, 12(3): 223.
[13] Agarwal U P, Reiner R S, Ralph S A. Cellulose, 2010, 17(4): 721.
[14] Agarwal U P, Reiner R S, Ralph S A. Journal of Agriculture and Food Chemistry, 2013, 61: 103.
[15] Agarwal U P, Ralph S A, Reiner R S. Wood Science and Technology, 2014, 48(6): 1213.
[16] Atalla R H, Dimick B E. Carbohydrate Research, 1975, 39(1): C1.
[17] Schenzel K, Almlöf H, Germgärd U. Cellulose, 2009, 16(3): 407.
[18] Jähn A, Schröder M W, Füting M, et al. Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy, 2002, 58(10): 2271.
[19] Schenzel K, Fischer S. Cellulose, 2001, 8(1): 49.
(Received Apr. 13, 2015; accepted Aug. 2, 2015)
*Corresponding author
Study on the Application of Raman Spectroscopy to the Research on Natural Cellulose Structure
MA Jian-feng, YANG Shu-min, TIAN Gen-lin, LIU Xing-e*
International Centre for Bamboo and Rattan, Beijing 100102, China
As the skeleton substances of lignocellulosic biomass cell wall and the precursor of biofuels production, the research on cellulose structure, an important natural biomarcromolecules, attracts great attention. Considering its in situ features and higher resolution, Raman spectroscopy has been used to investigate the structure of cellulose molecular chain and cellulose aggregation structure at multi-scale. In this paper, the configurations and corresponding parameters of two types of Raman spectroscopy (Dispersive Raman and FT-Raman) were compared. Subsequently, the utilization of Raman spectroscopy in cellulose micro-distribution, cellulose enzyme hydrolysis, cellulose chain orientation and deformation, cellulose crystallinity and polymorphic transformation was discussed in detail. Given the existing deficiencies of the Raman spectroscopy when used to investigate the natural cellulose, some suggestions were proposed in order to promote the application of Raman spectroscopy to the research of natural macromolecular.
Natural cellulose; Raman spectroscopy; Cellulose chain structure; Cellulose aggregation structure
2015-04-13,
2015-08-02
国际竹藤中心基本科研业务费项目(1632015002)和“十二五”国家科技支撑计划项目(2012BAD54G0103)资助
马建锋,1984年生,国际竹藤中心助理研究员 e-mail: majf@icbr.ac.cn *通讯联系人 e-mail: mjfxl31@126.com
TS01
A
10.3964/j.issn.1000-0593(2016)06-1734-06
猜你喜欢
杂志排行

光谱学与光谱分析的其它文章
- 基于光声光谱联合主成分回归法的血糖浓度无损检测研究
- Structural, Morphological and Optical Properties of Well-Ordered CdO Nanostructures Synthesized by Easy-Economical Chemical Bath Deposition Technique
- Sensitivity Enhancement in Uranium Determination by UV-Visible Spectroscopy Using Ion Imprinted Polymer
- 采用小波分析方法降低可调谐半导体激光吸收光谱技术测量下限的实验研究
- 钠钾替代条件下不同基因型棉花叶片的FTIR光谱研究
- 近红外高光谱成像技术用于转基因大豆快速无损鉴别研究