全基因组选择在植物育种中的研究进展
2016-07-03唐金梅陈建国
唐金梅,陈建国
(湖北大学生命科学学院,湖北武汉430062)
全基因组选择在植物育种中的研究进展
唐金梅,陈建国*
(湖北大学生命科学学院,湖北武汉430062)
育种值的估计是品种选育核心,在农业生产中占有十分重要的地位。全基因组选择通过估计全基因组所有标记或单倍型的效应,从而得到基因组估计的育种值,是分子标记辅助选择的一种新方法。随着高通量基因分型技术的发展及高密度全基因组SNP标记的开发应用,全基因组选择已成为动植物遗传育种的研究热点。对全基因组选择的原理、计算方法、影响准确性的因素及植物育种中的研究现状等进行综述,并对全基因组选择在植物育种的应用进行了展望。
全基因组选择;植物育种;研究进展
20世纪90年代始,分子标记辅助选择(MAS)已用于植物遗传改良,利用分子标记进行MAS育种可显著提高育种效率[1-2]。但MAS的应用有很大的局限性:从深度上看,世代数增加,重组率上升,从而引起分子标记-数量性状基因座(QTL)连锁的程度下降;MAS利用表型和部分基因信息进行遗传评估,但具有显著效应的基因或标记非常有限,这些基因或标记能解释的遗传变异也非常有限;找到的具有显著效应的相关标记QTL不能捕捉到微效基因[3]。从广度上看,MAS方法工作量大,发现并证实有效基因时间长,成本高,且1次只能研究极少量的QTL,而同时对不同性状的QTL间相互作用进行研究较难实现[4]。近年来,随着拟南芥、水稻和玉米等模式植物全基因组测序的完成[5-8],低成本高密度标记(SNP)标记的开发应用[9],动物育种领域的进展和生物信息学的迅猛发展,全基因组选择在植物育种中运用已成为研究热点。
1 全基因组选择的概念与原理
2001年,Meuwissen首次提出全基因组选择(genomic selection,GS)的概念[10]。GS是利用覆盖全基因组的SNP进行个体遗传评估,从而得到基因组估计的育种值(genomic estimated breeding value,GEBV)[8]。GS主要以连锁不平衡(linkage disequilibrium,LD)为基础,其基本理论是假设影响数量性状的每一个QTL至少与1个标记处于连锁不平衡状态[11],能够解释大部分的遗传方差[12]。
GS应用于动物育种领域时间已较长,但在植物育种领域尚处于初期[3]。全基因组选择是利用参考群体估计每个SNP的效应,然后利用SNP效应估计值计算育种群体的育种值(图1)。参考群体是由相关个体组成,每个个体均有性状表型记录和所有SNP基因型;育种群体通常是参考群体的后代或与参考群体相关的新品系,每个个体包含所有SNP基因型[3]。因此,全基因选择依赖于参考群体和育种群体的标记和位点连锁不平衡的遗传相似度[3]。
2 全基因组选择的方法
全基因组选择主要有2种方法,一是标记效应估计法,另一种是基于遗传关系矩阵预测GEBV的GBULP法。
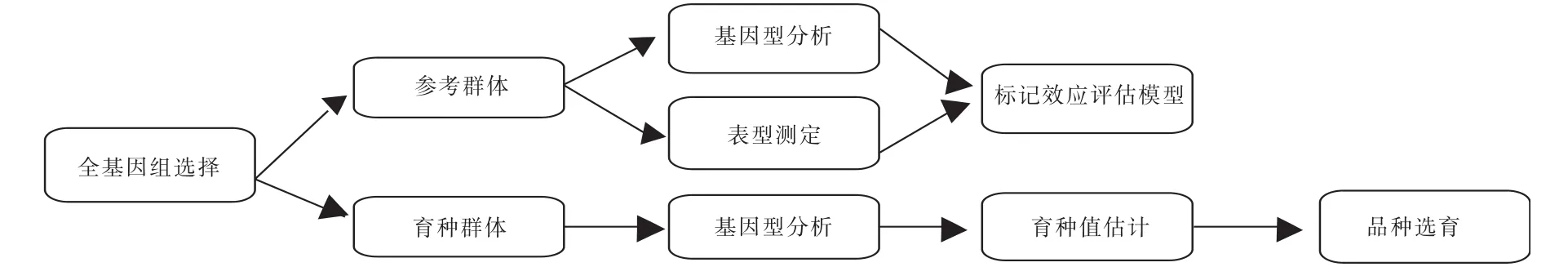
图示 全基因组选择流程Fig. Flow of genomic selection
2.1 标记效应估计法
建立标记效应评估模型:

其中,y是参考群体中个体的表型向量;(是总表型均值;xt是标记位点t的等位基因;(t是第t个标记的效应;e是随机残差,是残差方差。xt基因型矩阵可以用1、0和-1编码,分别代表基因型AA、AB和BB。
GEBV可以通过公式(1)得出:

其中,xnew是育种群体的个体等位基因矩阵;是回归系数αt的估计值。
全基因组选择中预测因子数(p)通常远远大于个体数目(n),在这种情况下普通最小二乘法的预测能力很低,因其将标记效应作为固定效应处理,导致预测因子的多重共线性和过度拟合。为克服此问题,得到更好的全基因组预测模型,现已提出多种方法,如岭回归最佳线性无偏估计(RR-BLUP)[7]、贝叶斯法(Bayesian)[7,1314]、偏最小二乘法(PLS)[15]、核回归和机器学习方法[16]等。
通过参考群体得到的预测模型需经交叉验证,从而得到最优拟合模型进行育种群体的GEBV评估。
2.2 基于遗传关系矩阵的GBULP法
GBULP法通过遗传关系矩阵预测GEBV,利用已测定的个体基因型计算其相关关系,记为G矩阵,并用G矩阵代替基于性状和系谱的最佳线性无偏预测(TP-BLUP)中的A矩阵,从而估计育种值[17]。
3 全基因组选择的准确性评估
GEBV预测的准确性是决定全基因组选择成功最重要的因素。预测的准确性rA表示GEBV与真实育种值(TBV)的相关度(Pearson correlation),一般来说准确程度越高,选择效果越好。rA可用以下公式计算:

式中,h2是狭义遗传力,Np是参考群体的个体数目,Me是独立染色体片段数,Me取决于有效群体大小(Ne)和以摩尔根表示的基因组长度(L):Me≈2 NeL。理想状况下,Me与有效QTL数相关,而h2和Np的结合是预测准确性的关键[18-19]。在低遗传力情况下,增加参考群体的个体数能保持模型预测的准确性[19]。
4 全基因选择模型准确性的影响因子
全基因组选择育种和其他育种方法相比具有较大优势,但全基因组选择的准确性易受统计模型效果、标记类型和密度、标记-QTL连锁不平衡的程度和分布、基因效应、遗传力及遗传结构、参考群体大小、参考群体与预测群体的世代距离和遗传距离影响[19]。
4.1 统计模型
全基因组选择模型准确性的差异主要在于各模型对标记效应方差分布的假设和处理不同。模拟研究发现,在全基因组选择的各种方法中,Bayes法和GBLUP法估计的准确性较高,LS法准确性最低[9]。目前研究表明,准确性为BayesB>BayesA>RR-BLUP[10,20]。BayesB法模拟数据中QTL数量有限,与BayesB法的理论假设比较吻合[21],故优于其他方法。
4.2 标记类型与标记密度
不同类型标记的多态信息含量不同,SNP标记等位基因具有二态性,其结果重复性好,故GS一般用SNP标记。增加标记密度可增加标记间连锁不平衡,有助于预测准确性的提高。有研究表明,当SNP标记密度从1Ne/morgan增加到8Ne/morgan,准确性可从0.69提高到0.86[22]。标记密度主要还由LD跨度和样本大小决定。玉米的LD跨度比大麦和小麦短,因此需更高的标记密度。用485个标记研究小麦发现,在达到最小标记数(128~256个)前,随着标记密度的增加预测准确性急剧增加;达到最小标记数时,准确性处于平衡状态;超过这个最小标记数后,准确性开始下降[23]。而玉米中用800个标记时准确性达到平衡[24],可见不同物种需要标记数不同。全基因组选择模型标记数目的确定依赖于目标群体有显著的LD标记区间。
4.3 标记-QTL连锁不平衡程度
连锁不平衡(LD)对GS操作也具有重要影响,标记和性状位点LD的缺失使后代育种准确性降低。LD是不同位点的等位基因非随机组合,2个位点的LD强度可用等位基因频率测定,一般用D、D′和r2表示。当D=D′=r2=0时,完全随机,无连锁不平衡;当|D|=0.25,即|D′|=r2=1时为完全连锁不平衡[25]。LD强度随着2个标记间距离的增大而下降。一般认为,当r2>0.1时LD强度显著。位点间等位基因频率和重组率、群体结构、群体大小、交配系统、遗传漂变及选择等因素均影响LD水平[11]。而这些影响因素中较重要的是突变和重组,突变能产生新的多态性,丰富遗传多样性;重组则能增加位点间的遗传多样性,使染色体内部的LD降低,一定程度上,LD的程度与重组率成反比。而对物种进行定向选择和驯化,从而形成对特定等位基因进行强烈选择,使该位点附近的遗传多样性减少,可增加该基因附近区域的LD水平。
4.4 遗传力及遗传结构
理想状态下,性状的遗传力越高预测的GEBV准确性越高,因随世代增加,有较高遗传力的性状其GS准确性降低较慢[14]。对于低遗传力的性状,在一定程度上增加表型记录世代数,可提高预测准确性,而高遗传力性状在表型记录世代数为2~3代时,GEBV准确性达最高并处于稳定状态。
4.5 参考群体大小
研究表明,增大参考群体规模可提高GEBV预测的准确性,由多个群体组成的参考群体优于由单一群体组成的参考群体。参考群体大小和标记基因型的平衡也很重要,若群体太小,基因型数据太大,易出现对基因型效应过度评估,即大p、小n的问题[26]。
研究表明,在进行GS研究时动物参考群体大于植物参考群体。主要有2个影响因素:第1个因素是植物种群的遗传多样性狭窄,主要是由自交繁殖或用于生成测试群体的亲本数量较少(经常使用双亲杂交)引起的。因更大的群体有丰富的遗传多样性,才能获得精确度高的GEBV[27],小的育种群体用于植物GS的研究,尤其是自交繁殖生产品种或双亲杂交衍生群体。第2个因素是大量留存的家系表型数据,这些表型数据降低了研究动物GS的成本。借鉴动物研究的方法,汇集植物种群的表型,从而满足高精度GEBV和低成本,实现植物GS研究。
4.6 参考群体与预测群体间世代距离及遗传距离
在实际育种工作中,新个体的加入让参考群体处于不断更新状态。GS效率每世代降低约5%,为保持GS长期优势,一般3~4代后必须重新对标记效应进行估计[10]。一般来说,参考群体和预测群体亲缘关系越近,GS准确性越高。
5 全基因组选择在植物育种中的应用研究
植物育种目标的多样性与不同物种的繁殖系统、基因组结构和利用的组织等有关。因此,GS应该应用各种选育方法,以适应不同植物的育种目标[28]。
GS运用于植物育种发展历程较短。2007年,Piyasatin等[29]首次将GS运用于植物,用自交系杂交模拟了GS选择的效率,但未考虑把特定的物种作为目标物种。Bernardo和Yu等[30]对特定物种玉米进行模拟研究,通过对3个周期的双单倍体品系(DHLs)的选择,对GS和分子标记轮回选择(MARS)进行比较显示,在QTL数量为20、40和100的情况下,GS的响应比MARS高18%~43%。除玉米的模拟研究外,还进行了2个GS模拟:远缘杂交种油椰子[31]和自花授粉作物大麦[32]。
除以上模拟双亲杂交群体外,有学者利用大麦自交系的真实基因型数据进行GS模拟研究,这些基因型数据主要来自单核苷酸多态性(SNP)与多样性阵列技术(DArT)[3335]。Lorenzana等[36-39]利用玉米、大麦、小麦和拟南芥对预测GEBV的准确性进行检验(表)。GS在动物领域比在植物领域的研究更广泛。动物育种群体差异性小,受环境因子影响较小,故GS在动物领域的适用性很强。Legara等[47]在小鼠中首次进行GS试验研究,到目前为止GS在奶牛育种中取得成就最显著。由表可见,在试验研究中虽然植物标记数比动物少,但植物中的准确性要高于动物中的准确性。表明,GS在植物育种领域的前景较好。

表 全基因组选择在植物和动物育种的预测能力Table Predictive ability of genomic selection in breeding of plants and animals
5 全基因组选择在植物育种中的应用展望
2006年底,荷兰Euribrid公司用20000个SNP标记对鸡进行育种值估计,这是全基因组选择运用的最早实例。之后加拿大、德国、美国、日本和墨西哥等相继对牛、猪、水稻和玉米等进行全基因组选择的研究。现在全基因组选择在动物育种领域和植物育种领域都取得了很大进展,但仍面临着较大的挑战。
GS应用的主要挑战有基因型与环境互作问题、群体结构问题、长期进行基因组选择的准确性问题、表型鉴定的准确性和成本问题、基因组选择可能导致稀有等位基因的丧失等。现在高密度的SNP标记在不断出现,故SNP效应估计的计算难度大大增加,对统计模型提出的要求更高。
目前,GS研究大部分集中在动物领域,并且取得了很大成就,虽然动物育种的目标、试验设计、育种程序和群体特性等与植物育种不同,但仍有诸多值得借鉴的研究方法。虽然植物全基因组育种还处于初期,但相信随着基因分型技术的发展和成本的降低,植物育种中的全基因组选择方法将不断完善,具有广阔的应用前景。
[1]Fernando R L,Grossman M.Marker assisted selection using best linear unbiased prediction[J].Genet Sel Evol,1989,21(4):467-477.
[2]Lander E S,Botstein D.Mapping mendelian factors underlying quantitative traits using RFLP linkage maps[J].Genetics,1989,121(1):185-199.
[3]Desta Z A,Ortiz R.Genome-wide prediction in plant improvement[J].Trends in Plant Science,2014,19 (9):592-601.
[4]田 郎.林木植物的全基因组选择[J].陕西林业科技,2015(4):115-131.
[5]Anon B.Analysis of the genome sequence of the flowering plant Arabidopsis thaliana[J].Nature,2000,408(6814):796-815.
[6]Yu J,Hu S N,Wang J,et al.A Draft Sequence of the Rice Genome(Oryza sativa L.ssp.Indica)[J].Science,2002,296(5565):1937-1942.
[7]Patrick S,Schnable.The B73Maize Genome:Complexity,Diversity,and Dynamics[J].Science,2009,326(5956):1112-1115.
[8]郭亮虎.植物全基因组选择育种研究进展与前景[J].山西农业科学,2015,43(11):1558-1562.
[9]Aluízio Borém,Roberto Fritsche-Neto.Biotechnology and Plant Breeding:Applications and Approaches for Developing Improved Cultivars[M].American:Academic Press;2014:105-133.
[10]Meuwissen T H,Hayes B J,Goddard M E.Prediction of total genetic value using genome-wide dense marker maps[J].Genetics,2001,157(4):1819-1829.
[11]吴永升.植物数量性状全基因组选择研究进展[J].西南农业学报,2012,25(4):1510-1514.
[12]Schaeffer L R.Strategy for applying genome-wide selection in dairy cattle[J].Animal Breeding and Genetics,2006,123(4):218-223.
[13]Solberg T R,Sonesson A K,Woolliams J A,et al.Genomic selection using different marker types and densities[J].Anim Sci,2008,86(10):2447-2454.
[14]Villumsen T M,Janss L,Lund M S.The importance of haplotype length and heritability using genomic selection in dairy cattle[J].Animal Breeding and Genetics,2009,126(1):3-13.
[15]Kotz S,Johnson N L.Encyclopedia of Statistical Science[M].New York:Wiley,1985:581-591.
[16]Long N,Gianola D,Rosa G J M,et al.Machine learning classification procedure for selecting SNPs in genomic selection:application to early mortality in broilers[J].Anim Breed Genet,2007,124(6):377-389.
[17]VanRaden P M.Efficient methods to compute genomic predictions[J].Dairy Sci,2008,91(11):4414-4423.
[18]Daetwyler H D.Ricardo P W,Beatriz R,et al.The impact of genetic architecture on genome-wide evaluation methods[J].Genetics,2010,185(3):1021-1031.
[19]Combs E,Bernardo R.Accuracy of genome-wide selection for different traits with constant population size,heritability,and number of markers[J].Plant Genome,2013,6(1):120.
[20]Lund M S,Sahana G,Koning D J,et al.Comparison of analyses of the QTLMAS XII common dataset.Genomic selection[J].BMC Proc,2009,3(S1):51-58.
[21]Calus M P L.Genomic breeding value prediction:Methods and procedures[J].Animal,2010,4(2):157-164.
[22]Solberg T R,Sonesson A K,Woolliams J A,et al.Genomic selection using different marker types and densities[J].Anim Sci,2008,86(10):2447-2454.
[23]Heffner E L,Jannink J,Iwata,et al.Genomic selection accuracy for quality traits in biparental wheat populations[J].Crop Sci,2011,51(6):2597-2606.
[24]Zhao Y,Gowda M,Liu W,et al.Accuracy of genome-wide selection in European maize elite breeding populations[J].Theor.Appl.Genet,2012,124 (4):769-776.
[25]Gaut B S,Long A D.The lowdown on linkage disequilibrium[J].The Plant Cell,2003,15(7):1502-1506.
[26]Jannin K,Lorenz A J,Iwata H,et al.Genomic selection in plant breeding:from theory to practice[J].Brief.Funct.Genomics,2010,9(2):166-177.
[27]Mujibi F D N,Nkumah J D,Durunna O N,et al.Accuracy of genomic breeding values for residual feed intake in crossbred beef cattle[J].Journal of Animal Science,2011,89(11):3353-3361.
[28]Varshney R K,Graner A,Sorrells M E.Genomicsassisted breeding for crop improvement[J].Trends in Plant Science,2005,10(12):621-630.
[29]Piyasatin N,Fernando R L,Dekkers J C M.Genomic selection for marker-assisted improvement in line crosses[J].Theoretical and Applied Genetics,2007,115(5):665-674.
[30]Bernardo R,Yu J.Prospects for genome-wide selection for quantitative trait in maize[J].Crop Science,2007,47(3):1082-1090.
[31]Wong C K,Bernardo R.Genome-wide selection in oil palm:increasing selection gain per unit time and cost with small populations[J].Theoretical and Applied Genetics,2008,116(116):815-824.
[32]Bernardo R.Genome-wide selection with minimal crossing in self-pollinated crops[J].Crop Science,2010,50(2):624-627.
[33]Zhong S,Dekkers J C M,Fernando R L,et al.Factors affecting accuracy from Genomic selection in populations derived from multiple inbred lines:a barley case study[J].Genitics,2009,182(1):355-364.
[34]Jannink J L.Dynamics of long-term genomic selection [J].Genet Sel Evol,2010,42(1):35-38.
[35]Iwata H,Jannink J L.Accuracy of genome-wide selection prediction in barley breeding programs:a simulation study based on the real single nucleotide polymorphism data of barley breeding lines[J].Crop Science,2011,51(5):1915-1927.
[36]Lorenzana R E,Bernardo R.Accuracy of genotypic value predictions for marker-based selection in biparental plant populations[J].Theor.Appl.Genet.2009,120(1):151-161.
[37]Zhao Y M,Mette M,Gowda C,et al.Bridging the gap between marker-assisted and genomic selection of heading time and plant height in hybrid wheat[J].Heredity,2014,112(6):638-645.
[38]Zhao Y M,Gowda M,Würschum T,et al.Dissecting the genetic architecture of frost tolerance in Central European winter wheat.[J].Exp.Bot,2013, 64:4453-4460.
[39]Albrecht T V,Wimmer V,Auinger H J,et al.Genome-based prediction of testcross values in maize [J].Theor.Appl.Genet,2011,123(2):339-350.
[40]Crossa J P,P rez P,Hickey J,et al.Genomic prediction in CIMMYT maize and wheat breeding programs[J].Heredity,2014,112(1):48-60.
[41]Hofheinz N D,Borchardt K,Weissleder M F.Genome-based prediction of test cross performance in two subsequent breeding cycles[J].Theor.Appl.Genet,2012,125(8):1639-1645.
[42]Zhao Y M,Gowda M,Liu W,et al.Choice of shrinkage parameter and prediction of genomic breeding values in elite maize breeding populations [J].Plant Breeding,2013,132(1):99-106.
[43]Zhao Y J,Zeng R,Fernando,J C R.Genomic prediction of hybrid wheat performance[J].Crop Sci.,2013,53(3):802-810.
[44]Guo T,Li H,Yan J,et al.Performance prediction of F1hybrids between recombinant in bred lines derived from two elite maize inbred lines[J].Theor.Appl.Genet,2013,126(1):189-201.
[45]Jacobson A L,Lian S,Zhong R,et al.General combining ability model for genome-wide selection in a biparental cross[J].Crop Sci,2014,54(3):895-905.
[46]Albrecht T H,Auinger V,Wimmer J O,et al.Genome-based prediction of maize hybrid performance across genetic groups,testers,locations,and years [J].Theor.Appl.Genet,2014,127(6):1375-1386.
[47]Legarra A,Robert-Granie C,Manfredi E,et al.Performance of genomic selection in mice[J].Genetics,2008,180(1):611-618.
[48]Hayes B J,Bowman P J,Chamberlain A J,et al.Genomic selection in dairy cattle:progress and challenges[J].Journal of Dairy Science,2009,92(2):433-443.
[49]Luan T,Woolliams J A,Lien S,et al.The accuracy of genomic selection in Norwegian red cattle assessed by cross-validation[J].Genetics,2009,183(3):1119-1126.
[50]Wolc A,Stricker C,Arango J,et al.Breeding value prediction for production traits in layer chickens using pedigree or genomic relationships in a reduced animal model[J].Genetics Selection Evolution,2009,43 (1):5-8.
(责任编辑:刘忠丽)
Progress of Genomic Selection in Plant Breeding
TANG Jinmei,CHEN Jianguo*
(Faculty of Life Science,Hubei University,Wuhan,Hubei 430062,China)
Breeding value estimation is the core of variety development,which plays an important role in agricultural production.Genomic selection is a new approach of molecular marker assisted selection by estimating the effects of all the markers or haplotypes in the whole genome.This method can be used to improve the accuracy of breeding value estimation,shorten the generation interval,facilitate earlier selection,accelerate genetic progress and reduce the cost.With the development of high throughput genotyping technology and the advent of high density SNP markers,genomic selection has become a hot spot in the research of animal and plant genetics and breeding.The authors summarized the principles,methods,factors affecting the accuracy of genomic selection,and the research status and problems in plant breeding.
genomic selection;plant breeding;research progress
Q943.2
A
1001-3601(2016)08-0326-0001-05
2015-12-16;2016-07-26修回
湖北省农科院粮食作物种质创新与遗传改良湖北省重点实验室开放项目“早稻恢复系R287持绿性的遗传研究及作图群体构建”(2014lzjj05)
唐金梅(1988-),女,在读硕士,研究方向:分子植物育种。E-mail:594133857@qq.com
*通讯作者:陈建国(1966-),男,教授,从事植物数量遗传研究。E-mail:jgchen@hubu.edu.cn
