基因编辑的发展历程
2016-05-30郭晓强
郭晓强
基因编辑从传统的基因打靶,到改进的嵌合核酸酶技术,再到最新的RNA或DNA指导的核酸酶技术,取得了快速发展,为生命科学研究和临床应用提供了简捷有效的操作工具。其应用潜力巨大,但仍须解决脱靶等相关技术问题。
DNA是遗传信息载体,基因是一段具特定生物学功能的DNA片段,决定生物体生老病死等各种生命过程。基因编辑(gene editing)指根据科研或临床实际需要对目的基因(亦称靶DNA)进行插入、移除或替换等精确遗传操作,从而达到预定目的(引入或修复突变)之过程。鉴于基因在正常发育和疾病发生中的重要性,这项研究的意义不言而喻。而要完成基因编辑,工具必不可少,多种核酸内切酶的发现为各种技术的发明提供了可靠保证。在此基础上基因编辑技术经历了多阶段的发展过程并显示了多方面的应用前景。
限制性内切酶的发现与应用
故事要从1950年代讲起。1952年,意大利裔美国微生物学家卢里亚(S.E.Luria,1969年诺贝尔生理学或医学奖获得者)发现了细菌限制一修饰现象。同一种噬菌体对不同菌株具有不同的感染力,如极易感染大肠杆菌C菌株的入噬菌体对K菌株感染力仅是对C菌株的千分之一,因此将大肠杆菌K菌株抵抗入噬菌体感染称为“宿主控制的噬菌体限制”,但对这种现象的机制缺乏了解。
1962年,瑞士科学家阿尔伯(W.Arber)提出解释细菌修饰一限制机制的假说。他推测细菌内存在两类酶,一类为核酸内切酶,可特异识别噬菌体DNA并进行剪切,从而限制入侵;另一类为DNA甲基化酶,对自身DNA相应序列进行甲基化修饰,破坏核酸内切酶的自我识别,从而避免“误伤”。
1968年,阿尔伯和美国梅塞尔森(M.S.Meselson)同时从大肠杆菌鉴定出核酸内切酶,该酶可识别特定DNA序列,但是在随机位置上剪切DNA,不具特异性,称为Ⅰ型限制性内切酶。
1970年,美国微生物遗传学家史密斯(H.O.Smith)和同事又从嗜血流感细菌(Haemophilusinfluenzae)中分离得到一种核酸内切酶,该酶可特异识别噬菌体双链DNA并发挥剪切作用,但对细菌DNA无效,符合阿尔伯当初的预测。史密斯和同事还鉴定出该酶的识别及剪切序列。该酶被命名为HindⅡ,这是人类发现的第一个Ⅱ型限制性内切酶。HindⅡ的发现引起科学界极大关注,从而掀起了一场寻找Ⅱ型限制性内切酶的热潮。另一方面,限制性内切酶的巨大应用潜力不久就被史密斯同事内森斯(D.Nathans)发现和首先开发。
1970年,内森斯利用史密斯赠予的限制性内切酶HindⅡ对猿猴病毒40(SV40)环形DNA进行酶切,获得大小不同的11个DNA片段(意味着SV40基因组含有10个HindⅡ的识别与剪切位点),首次获得了SF40基因组的限制性内切酶图谱。
1978年,阿尔伯、史密斯和内森斯因“限制性核酸内切酶的发现及分子遗传学应用”而分享诺贝尔生理学或医学奖。限制性内切酶的发现及应用为DNA研究提供了重要工具,它们被称为“DNA操作的手术刀”。
DNA重组和基因工程
在内森斯切开SV40 DNA基础上,美国斯坦福大学生物化学家伯格(P.Berg)又往前迈进了一大步。
1972年,伯格在体外用限制性内切酶将SV40DNA和噬菌体DNA分别切开,嗣后又借助DNA连接酶将它们重新连接为整体,从而首次在体外实现了两个不同来源DNA的人工重组,伯格因这项成就分享1980年诺贝尔化学奖。不久,科恩(S.N.Cohen)和博耶(H.Boyer)进一步把重组DNA转入大肠杆菌,开启了基因工程大门。重组DNA和基因工程的成功,推动了生物技术领域的快速发展,更重要的是给科研人员提供了无限想象空间,即人类可以用技术手段对DNA进行主动操作,这奠定了今天基因编辑的思想基础。
同源重组与基因打靶
最早的基因编辑利用机体自身的同源重组(homologous recombination)。同源重组是指两段相似或相同序列的DNA发生交换的现象,主要用于DNA双链断裂后修复,此外还是减数分裂期新序列产生的基础。早在1947年,美国微生物学家莱德伯格(J.Lederberg。1958年诺贝尔生理学或医学奖获得者)就在细菌中发现了遗传重组现象,1970年代在哺乳动物中也鉴定成功,但对于机体内部的同源重组是否可应用于人工遗传操作,人们不得而知。
1980年代初,意大利裔美国遗传学家卡佩基(M.R.Capecchi)坚信同源重组可应用于细胞内DNA的操作。他首先获得一种携带新霉素抗药基因缺陷的细胞系(添加新霉素可造成细胞死亡),然后通过显微注射方式将正常新霉素抗药基因导入细胞,结果细胞恢复了抗性。实验也证实,通过同源重组方式可完成基因替换。与此同时,另一位美国科学家史密西斯(O.Smithies)完成了珠蛋白的同源重组实验。1987年,多家实验室在小鼠胚胎干细胞上利用同源重组实现基因替换和引入突变,并最终制备成功疾病模型。这种技术一般称为基因打靶(gene targeting)。随后经过完善与改进,该技术在生命科学多个领域中得到广泛应用,卡佩基、史密斯以及另外一位科学家也因此分享2007年度的诺贝尔生理学或医学奖。
虽然基因打靶技术取得巨大成功,但是该技术存在一个明显缺陷,就是在正常情况下,哺乳动物细胞和模式动物体内的同源重组发生率极低,因此基因打靶技术成功率不高。后来发现,DNA双链断裂(double-strand break,DSB)可使同源重组效率大大提高,于是研究人员开始寻找增加细胞内DSB发生的策略,他们的眼光重又回到限制性内切酶。
大范围核酸酶
然而,传统的限制性内切酶也存在一个巨大不足,其大部分识别位点在4~6个碱基对(base pair,bp)。这对体外短片段DNA的操作问题不大,但对基因组层面的操作则问题多多。以剪切6bp的内切酶为例,理论上平均4096(46)个碱基就存在一个剪切位点。如果将这类酶应用于人基因组(30亿碱基对)的编辑,理论上一次可产生7.3×105个以上断裂点;若要保证切割位点唯一性,则须识别16个碱基(416≈43亿)以上(随后发展的多种基因编辑技术的常见识别位点在20个上下),传统限制性内切酶显然无法应用。幸运的是,自然界还存在一类特殊的核酸内切酶。
大范围核酸酶(meganuclease)亦称归巢核酸内切酶(homing endonuclease),存在于原核生物和真核生物的线粒体/叶绿体等当中,大部分由内含子编码。大范围核酸酶可特异识别DNA长片段,一般在12~40bp之间,在数量上能满足需求。1985年,一种大范围核酸酶I-SceI被发现,它可特异性识别18 bp的DNA片段,在此基础上将DNA切开而产生双链断裂。1990年代,I-SceI被用于辅助基因编辑,可使传统基因打靶效率大大提高。
但大范围核酸酶在基因编辑的应用方面有重大瓶颈。首先,天然大范围核酸酶种类有限,对于人类大部分基因找不到相应的酶识别;而新找一个识别给定序列的大范围核酸酶,其机会实在太渺茫。其次,改进的余地也有限。大范围核酸酶的识别与剪切两种功能利用同一结构域,改造识别结构域就会影响剪切活性,因此最终找到理想的酶是一项几乎无法完成的任务。科学家必须寻找新的策略。
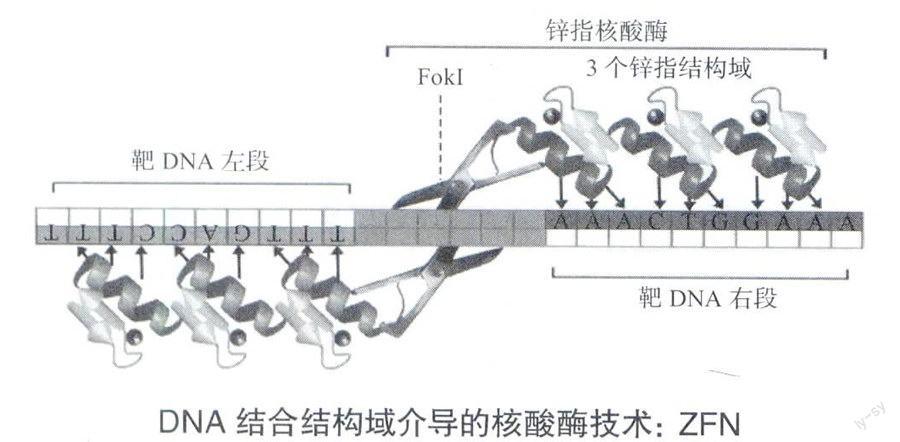
锌指核酸酶(ZFN)
1981年从海床黄杆菌(Flavobacteriumokeanokoites)中分离得到一种传统的IIS型限制性内切酶,被命名为FokI。相对于其他限制性内切酶,FokI拥有自己独特的结构,含两个相对独立的结构域,一个是N端的DNA结合结构域(可特异识别5-GGATG-3),另一个为c端的DNA剪切结构域。此特征为工程化改造提供了良机。因识别序列太短(仅5bp),天然FokI酶自然也无法应用于基因编辑,但相对独立的剪切结构域倒是可以作为DNA“剪刀”,跟具有识别功能的其他结构域联合使用。
转录因子是一类特异识别DNA序列并促进基因转录的蛋白质。转录因子识别DNA主要通过三类结构域来完成,其中一类为依赖锌离子的锌指结构域(zinc finger),典型特征是大约30个氨基酸可识别3个核苷酸,最终使64种可能的核苷酸三联体都有相应的锌指结构域加以识别。研究人员可根据需要将这些锌指结构域进一步联合,制造出一系列识别3的倍数核苷酸的组合体,如识别18个核苷酸理论上需6个锌指结构域串联即可,这种特征使它们成为DNA特异识别之重要工具。
1996年,美国约翰·霍普金斯大学钱德拉西格兰(S.Chandrasegaran)首次将三个串联的锌指结构域(识别9个核苷酸)与FokI的C端内切酶结构域通过一段连接蛋白融合,制造出第一个嵌合型核酸内切酶——锌指核酸酶(zinc finger nuclease,ZFN),并在体外证明该酶对靶DNA具特异剪切能力。此外,ZFN发挥活性时往往采用同源二聚体方式,特别是FokI核酸内切酶结构域必须在两端DNA均完成识别的前提下方可执行剪切作用,从而最大限度地避免了脱靶效应的发生。
随后研究人员探索这种嵌合核酸酶是否也可在细胞内对靶DNA起识别与剪切作用。2001年,首次在果蝇体内尝试成功,与传统基因打靶技术联用可显著增加基因编辑效率,迅速成为基因编辑领域的新热点。
ZFN技术相对于大范围核酸酶而言是巨大进步,但依然存在许多不足,其中针对DNA特异性序列的锌指结构域的组合与设计是极大挑战,导致技术进展缓慢,虽取得一定成功,但很难在大部分实验室里普及。
转录激活样效应蛋白核酸酶(TALEN)
许多革兰氏阴性菌在感染宿主过程中,可将自身蛋白质注入细胞,通过影响特定基因的表达而增强自身的生存适应性。2007年德国哈勒一维腾贝格大学(University of Halle-Wittenberg)的博纳斯(U.Bonas)发现植物病原菌黄单胞菌属(Xanthomonasgenus)可制造一种“毒力”蛋白质,该蛋白质由于具有激活宿主基因表达的功能,被命名为转录激活样效应蛋白(transcription activator-like effector,TALE)。2009年,博纳斯以及美国艾奥瓦州立大学的波格丹诺夫(A.Bogdanove)同时阐明了TALE激活基因表达的作用机制。TALE的结构由三部分组成,分别为核定位序列、DNA识别结构域和靶基因转录激活结构域。TALE的DNA识别结构域由多个单体串联重复构成,每个单体均含34个氨基酸,除12和13位氨基酸外其他序列完全相同,而正是这两个氨基酸的差异决定了核苷酸识别的特异性。每一个TALE识别单体对应于一种核苷酸,因此DNA四种碱基只需四种单体即可(远少于ZFN的64种结构域),从而使得构建多核苷酸识别的设计大大简化了。
随后,研究人员将根据靶DNA序列设计的TALE单体串联后,也通过连接蛋白与FokI的c端内切酶结构域相连,构建出TALE核酸酶(TALE nuclease.TALEN)。体内外实验表明,它可以实现对靶DNA的特异性剪切,迅速应用于基因编辑领域,并先后构建成多种模型动物,成为一颗耀眼新星,被《自然方法学》杂志评为“2011年度方法”。
TALEN相对于ZFN的高效、简洁,获得许多科研人员青睐。尽管在TALE单体的串联设计方面仍存在诸多不确定性,但他们相信,问题可以通过进一步完善技术得到解决。就在大家对TALEN技术的前景充满期许时,另一高效基因编辑技术横空出世,瞬间改变了该领域发展走向。
RNA介导的DNA编辑(CRISPR-Cas9)
1987年一个日本团队在细菌DNA中首次意外发现一种特殊结构,其中“重复序列一居间序列(spacer)-重复序列”交替出现。随后发现此特殊结构在原核生物中普遍存在。2002年研究人员将其统称为成簇规律性间隔短回文重复(clustered regularlyinterspaced short palindromic repeat,CRISPR);并将序列附近的编码基因命名为CRISPR相关因子(CRISPR-associated,Cas),推测它们参与CRISPR生物学功能。
2005年,研究发现CRISPR居间序列来自噬菌体或质粒等细菌染色体以外的DNA,这一发现暗示了它们重要的生理功能。在借鉴真核生物RNA干扰的基础上,提出了解释CRISPR-Cas系统作用机制的获得性免疫假说:CRISPR序列可转录出包含居间序列的RNA,该RNA与外源DNA转录出的RNA互补形成双链结构,随后Cas蛋白对外源RNA进行剪切,从而达到抵御病毒入侵的目的。
2007年,法国丹尼斯克(Danisco)集团微生物学家巴朗古(R.Barrangou)等以嗜热链球菌为材料证明CRISPR-Cas确是一种细菌获得性免疫系统。这一突破立刻引起许多科学家高度重视,因此想进一步探索其作用机制。2008年,两个研究小组发现CRISPR.Cas系统在发挥免疫机制的作用时确实可转录出RNA,即crRNA(CRISPR-related RNA)。crRNA有引导作用,然而它们更多识别的是DNA,而非当初假说认为的RNA。Cas蛋白有核酸内切酶活性,多种Cas蛋白联合可特异性地剪切与crRNA互补的DNA而形成双链断裂。该发现重要性显而易见,crRNA负责识别,Cas蛋白负责DNA剪切,两者具有用于基因编辑的两项基本能力。不过已发现的两类CRISPR-Cas系统(Ⅰ型和Ⅲ型)均较复杂,特别是进行DNA剪切时需多个Cas9蛋白共同作用,而ZFN和TALEN都只需一种嵌合核酸酶即可,因此应用优势并不明显。
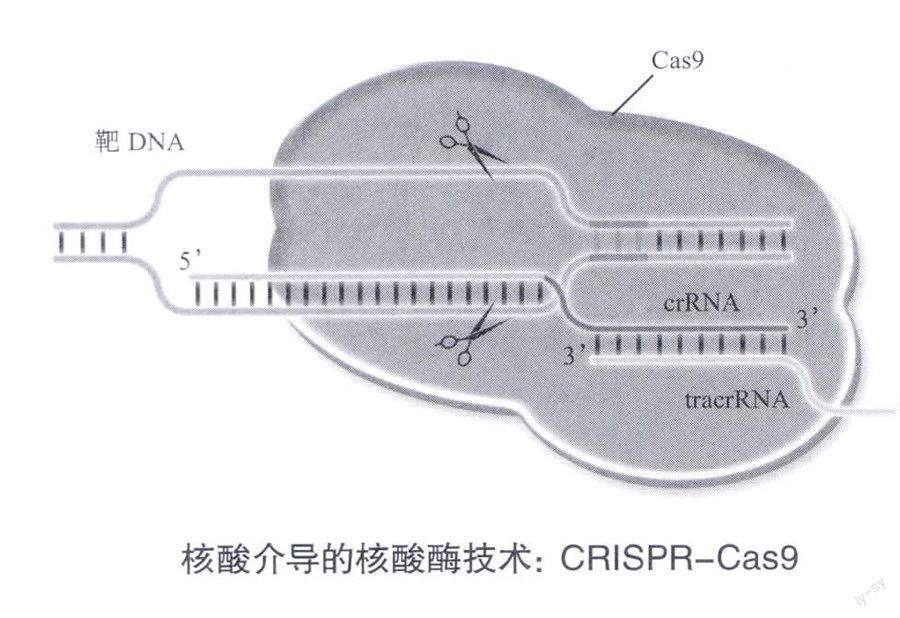
2011年,瑞典于默奥大学(Umefi University)微生物研究中心法国微生物学家沙尔庞捷(E.M.Charpentier)和同事在研究Ⅱ型CRISPR-Cas系统时发现,crRNA参与DNA识别还需反式激活的CRISPR来源的RNA(trans-activating CRISPR-derived RNA,tracrRNA)协助。这虽会增加RNA的需求数量(而RNA操作相对容易),但执行DNA剪切时只需一种Cas9蛋白即可。此新型CRISPR-Cas系统还吸引了美国加州大学伯克利分校结构生物学家杜德娜(J.A.Doudna),她与沙尔庞捷共商改造Ⅱ型系统。
2012年,杜德娜和沙尔庞捷联合小组将crRNA和tracrRNA两种RNA二合一,形成单链引导RNA(single guide RNA,sgRNA),其中crRNA部分负责与靶DNA配对识别,而tracrRNA负责维持空间结构,在RNA指导的核酸酶(RNA-guided nuclease,RGN)Cas9催化下,完成对靶DNA双链的特异剪切。2013年,哈佛大学的张锋和丘齐(G.M.Church)研究小组采用这套系统,在细胞内完成靶基因编辑。
CRISPR-Cas9相对于传统的嵌合核酸酶技术,优点显而易见。首先是设计简单,摈弃了嵌合酶技术的蛋白质结构域DNA识别设计理念,随之用符合沃森一克里克配对的理念进行sgRNA设计。其次在操作方面,传统嵌合酶设计在识别结构域后还要与核酸内切酶剪切结构域连接,如果有多个靶DNA的编辑,就需要多次的融合。CRISPR-Cas9在Cas9保持不变前提下只需要对sgRNA进行操作,因此在同时进行多基因编辑方面更显高效。CRISPR-Cas9由于具有高效简便的特点,一经发现便引发了一场基因编辑研究的热潮,这项技术迅速风靡全世界。
DNA介导的DNA编辑(ssDNA-Ag0)
1998年,美国法尔(A.Z.Fire)和梅洛(C.C.Mello)在合作研究线虫肌肉相关蛋白质的生物学功能过程中,发现同时注入针对该基因的正义RNA和反义RNA能起干扰基因表达的作用,此现象被称为RNA干扰(RNA interference,RNAi)。2006年,法尔与梅洛为此分享诺贝尔生理学或医学奖。
RNA干扰现象的发现,激发了随后对其作用机制的研究。原来,双链RNA首先被加工为21~23个核苷酸的双链RNA,然后其中一条链与靶mRNA及相关因子形成RNA诱导的沉默复合体(RNA-inducedsilencing complex,RISC),完成对mRNA的剪切,而Argonaute(Ago)蛋白是RISC的一种关键成分。
Ago蛋白最初在模式植物拟南芥中发现,由于基因突变体形态非常类似于软体动物船蛸(Argonautaargo)而得名。随后从果蝇、小鼠和人等发现Ago普遍存在。Ago蛋白拥有一个C端Piwi结构域,具RNA核酸内切酶活性,从而对靶mRNA实现剪切。进一步研究发现,不仅真核生物而且包括细菌和古菌在内的原核生物亦普遍存在Ago蛋白。借鉴CRISPR.Cas9研究经验,研究人员提出,原核生物Ago蛋白也参与细菌免疫机制,意味着也能用于基因编辑。
2014年,荷兰瓦赫宁根大学(WageningenUniversity and Research Centre)范德奥斯特(J.van der Oost)发现古菌——噬热栖热菌(Thermusthermophilus)的Ago蛋白(TtAgo)还具有DNA核酸内切酶活性,但和Cas9不同,TtAgo剪切靶DNA时依赖单链DNA(single stranded DNA,ssDNA)而不是RNA,说明Ago蛋白是一种DNA指导的核酸酶(DNA-guided nuclease,DGN)。此发现在阐明一种新型原核生物的免疫系统作用机制同时,还显示ssDNA-Ago系统也可应用于基因编辑。然而,该技术的应用前景尚待进一步确定。鉴于三大类生物分子DNA、RNA和蛋白质的生物学作用特征,即DNA是遗传信息携带者,蛋白质是生物学功能具体执行者,而RNA更多发挥中介和调节等多重生物学作用(近几年生命科学进展更多出现在RNA领域,包括核酶、RNA干扰等),功能多样的RNA作为引导分子,可能比DNA在基因编辑方面具有优势。当然,此推测需要充分的实验加以证实。
基因编辑的应用
基因编辑工具更多地引入DNA双链断裂(DSB),由于机体对DNA完整性的要求,因此随后会启动修复机制。DNA修复系统主要有两种类型,分别为非同源末端连接(non-homologous end ioininz.NHEJ)和同源指导的修复(homology-directedrepair,HDR)。NHEJ无需同源序列,可将断裂的双链重新连接,然而在连接过程中会出现碱基丢失或增加,最终造成基因的缺失或插入突变。HDR是在同源DNA存在前提下,以同源DNA信息为模板完成DNA断裂处修复。如果按基因打靶策略在引入双链断裂的同时补充打靶序列,可实现精确替换,既可引入定点突变,又可实现突变基因的修复。基因编辑通过对目标DNA的精确操作,达到控制生物性状和行为的目的,因此拥有广泛的应用前景。
首先,可作为便捷的实验工具。实验室基因功能研究的快捷方法是RNA干扰技术,但RNA干扰只影响mRNA含量,并未去除基因本身,而传统基因敲除由于费事费钱,其应用受到限制。当前的基因编辑技术可快速制备突变体,加快了实验研究进程。
其次,用于模式生物制备。用传统基因打靶技术制备的模式动物,一方面效率低,另一方面还受限于干细胞;而用最新基因编辑技术已完成大量物种的基因敲除,制备出各种特定疾病的动物模型,并且时间大大缩短。采用CRISPR-Cas9等技术不仅能对传统模式动物如小鼠、大鼠等进行基因编辑,还能在大型哺乳动物如猪和牛以及猴等灵长动物中开展基因编辑。
第三,用于物种改良。新的基因编辑技术对动植物改良有重要意义,如借助CRISPR-Cas9完成对猪体内病毒的去除工作等多方面的应用。
第四,具有潜在的临床价值。最新的基因编辑技术已在遗传病、血液病和感染病等领域显示了巨大临床价值,还为其他疾病的诊治提供了重要借鉴。当然尚需进一步的探索以保证应用的安全性。
第五,用于其他领域。通过对现有技术的改造,可望在基因表达调控等领域也发挥重要作用。
基因编辑的前景
基因编辑从原理上讲只有一种类型,就是借助核酸酶实现DSB,但根据DNA序列特异识别机制的差异,可分为两大类,即嵌合酶技术(如ZFN和TALEN等)和核酸指导的核酸酶技术(CRISPR-Cas9和ssDNA-Ago等)。基因编辑的演变历史也基本沿着细菌-免疫系统-核酸酶这一线索展开。
由基因编辑发展历程可见科学发展一般规律。第一代基因编辑为单组分(天然核酸内切酶或人工嵌合核酸酶),第二代基因编辑为双组分(引导核酸加核酸内切酶),突破性进展在于用精确碱基配对取代蛋白质一DNA识别。但两代技术均存在一个重大缺陷,就是需要原核核酸内切酶,意味着应用于临床存在诱发机体免疫应答的潜在危险。因此推测,是否有诞生第三代基因编辑的可能?即重回单组分,但此时已不是蛋白质而是RNA。RNA本身既具有识别作用,有时又具有核酸内切酶活性(核酶),这样既减少了多组分困扰,又避免了外源蛋白质(对已有基因编辑技术必不可少)的不利影响。当然,这想法能否实现还要深入研究。
基因编辑研究分四个层面,即体外实验、细胞水平、动物水平和临床应用。目前研究人员已在前三个层面取得重大突破,对其临床应用也充满期待。然而。真要把基因编辑应用于临床,目前仍有许多问题要解决,如怎样提升体内基因编辑过程中的转染和编辑效率,怎样避免潜在的脱靶效应及不确定因素影响,此外还涉及伦理问题,须进一步完善相关技术,尽可能避免不良效应。上面提到DSB的引入,这也意味着正常情况下细胞内DSB发生率极低,暗示引入过多DSB对细胞可能是颇大威胁,为基因编辑安全性埋下了隐患。
总之,基因编辑技术犹如双刃剑,我们在看到其快速发展和广阔应用前景同时,也要理性估计其应用的得失,尤其对未来可能的临床应用更宜采取慎重态度。
关键词:基因编辑 基因打靶 锌指核酸酶 TALEN CRISPR-Cas9 ssDNA-Ago
