中国成人自身炎症性疾病临床和基因型特点:成人自身炎症性疾病单中心报道
2016-04-11曾小峰
沈 敏,吴 迪,曾小峰
(中国医学科学院 北京协和医学院 北京协和医院风湿免疫科 风湿免疫病学教育部重点实验室, 北京 100730)
ChinJAllergyClinImmunol,2016,10(4):325- 333
自身炎症性疾病(autoinflammatory diseases,AUID)是在2000年左右被定义的一组疾病,由于基因突变使其编码蛋白发生改变,导致固有免疫失调而引起全身炎症反应[1]。与自身免疫性疾病不同的是,AUID通常缺乏自身抗体或抗原特异性T细胞,其参与炎症损伤过程的主要是单核巨噬细胞,而非T、B细胞[2]。临床上AUID常常表现为复发性全身性炎症,包括突发周期性发热,伴皮疹、浆膜炎、淋巴结肿大和关节炎等,发作期急性期反应物升高;而发作间期,患者临床症状消失,急性期反应物也完全正常。
AUID主要包括一组单基因遗传病,如家族性地中海热(familial mediterranean fever,FMF)、甲羟戊酸激酶缺乏症(mevalonate-kinase deficiency,MKD)、肿瘤坏死因子受体相关周期性综合征(tumor necrosis factor-receptor associated periodic syndrome,TRAPS)、冷炎素相关周期性综合征(cryopyrin-associated periodic syndrome,CAPS)、NLRP12相关自身炎症性疾病(NLRP12-autoinflammtory disease,NLRP12-AD)、Blau综合征(Blau syndrome,BS)等[3- 4]。AUID还包括一些多基因疾病,如周期性发热-阿弗它口炎-咽炎-淋巴结炎(periodic fever, aphthous stomatitis, pharyngitis and adenitis,PFAPA)综合征、成人Still病(onset Still disease,AOSD)、全身型幼年特发性关节炎等[3- 4],其中PFAPA综合征发病机制不详,目前也未发现任何与之有关的致病基因[5- 6]。
AUID由于其遗传性特点大多发病较早,从出生后数小时到十多岁青少年期均可发病[7],部分患者成年后才发病,还有许多患者虽然幼年时发病,但直至成年后才获得确诊[8- 11]。由于AUID是一组相对少见而新的疾病,临床医师往往对其认识不足。此外,与儿童起病的AUID相比,成人起病的AUID往往临床表现更不典型,基因型以低外显基因突变更多见[12- 13],这使得成人AUID诊断更加困难。本研究对中国汉族成人AUID的临床特点和基因型进行分析,并与国外成人AUID中心的报道进行比较。
对象和方法
研究对象
从2015年4月至2016年5月,北京协和医院(Peking Union Medical College Hospital,PUMCH)风湿免疫科成人AUID中心(以下简称PUMCH中心)前瞻性纳入拟诊AUID的成年患者。拟诊AUID者需符合2010年Kastner教授定义[14],即易感宿主体内出现由固有免疫系统异常介导的异常增强的炎症,主要表现为周期性发热、皮疹、关节炎/痛、肌痛、淋巴结肿大、急相反应物质升高等,发作间期症状缓解,无高滴度自身抗体,并可排除感染、肿瘤等疾病。
纳入及排除标准
本研究仅纳入年龄≥16岁的患者。所有单基因AUID的诊断需结合临床表现、基因检测阳性结果和对治疗的反应。PFAPA综合征诊断依据目前达成共识的诊断标准(除起病年龄<5岁以外)[15]。
排除已确诊其他多基因和多因素AUID疾病,如白塞病、AOSD、克罗恩病患者,因为这些患者进行基因检测的原因并非是拟诊某种单基因AUID。如果患者临床表现符合拟诊AUID,但基因检测未发现与单基因AUID致病基因相符的结果,并能够进一步除外感染、肿瘤和其他自身免疫性疾病,且对传统抗炎药物或生物制剂治疗反应良好者,则诊断未分化AUID。未分化AUID未纳入本研究最终的数据结果分析。
基因检测结果比对Infevers网站(http://fmf.igh.cnrs.fr/ISSAID/infevers/)(目前公认的AUID基因变异数据库),并按照新近发表的AUID基因诊断指南[16],将检测到的基因变异分为3类:(1)明确的致病性基因变异(常常与较严重的临床表型相关);(2)低外显或不完全外显基因变异(常常与较轻的临床表型相关,也可见于正常人群或一级亲属);(3)单核苷酸多态性(通常被认为是非致病性基因变异,但并不总是)。
研究方法
收集所有成人AUID(单基因AUID和PFAPA综合征)患者的临床资料,包括年龄、性别、起病年龄、诊断年龄、主要临床表现、随访时间、治疗方案及对治疗的反应。并对所有患者进行包含单基因AUID致病基因在内的全基因组二代测序。数据采用均数或中位数(范围)的方法描述。并将本中心的结果与国外其他成人AUID中心的研究结果进行比较[13,17- 21]。
结 果
PUMCH中心成人AUID总体情况
自2015年4月至2016年5月共1年时间内,PUMCH中心共纳入37例拟诊AUID患者,并全部进行基因检测。16例(43.2%)患者基因检测阳性而确诊单基因AUID,另有2例诊断PFAPA综合征(基因检测结果阴性)。最终,共18例成人AUID患者在本中心获得确诊并随访,占所有拟诊AUID患者的48.6%(图1)。
这18例成人AUID患者包括FMF 7例(38.9%),TRAPS 2例(11.1%),CAPS 3例(16.7%),NLRP12-AD 3例(16.7%),BS 1例(5.6%)以及PFAPA综合征2例(11.1%)。18例患者均为汉族,男性较多见(男∶女= 13∶5)。平均起病年龄(25±16)岁(0~50岁),其中幼年起病者仅3例(16.7%),其余15例(83.3%)均成年起病。全部患者均在成年后方获得诊断,诊断时平均年龄为(34±10)岁(18~52岁)。从起病至最终确诊时间(延迟诊断时间)平均长达(10±9)年(0~29年)。本组患者除1例BS患者外,均无AUID家族史。
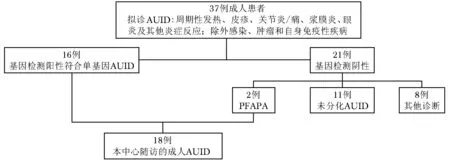
图1本研究成人AUID诊断流程图
Fig1AUID diagnostic flow chart for adults in this study
AUID:自身炎症性疾病
拟诊AUID的37例患者中21例(56.8%)基因检测结果阴性,即未发现与单基因AUID致病基因相符的基因突变(图1)。除外2例诊断PFAPA综合征者,尚有11例诊断未分化AUID。其余8例(21.6%)基因检测结果阴性的患者最终诊断为其他疾病,包括幼年特发性关节炎全身型3例(8.1%),AOSD 2例(5.4%),结节性多动脉炎、中枢神经系统血管炎和白塞病各1例(各2.7%)。
PUMCH中心与国外成人AUID中心结果比较
对比本中心与国外其他成人AUID中心研究结果发现(表1),成人AUID以FMF最常见,其次是CAPS或者TRAPS。本中心TRAPS所占比例较国外报道低,而NLRP12-AD比例高于国外中心的数据。成人MKD和BS患者均罕见。
PUMCH中心成人AUID的临床特点
18例成人AUID患者的主要临床表现见图2及表2。
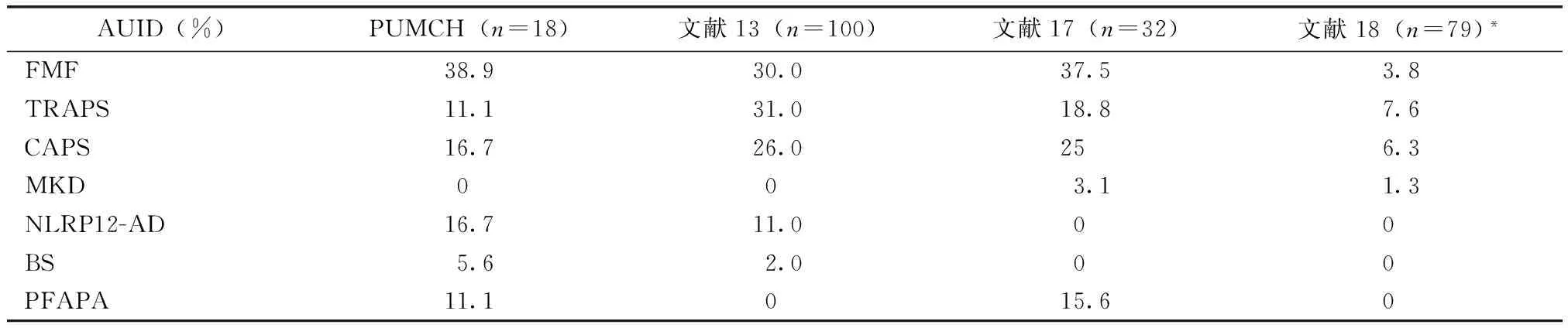
表1 北京协和医院成人AUID中心临床资料与国外其他中心比较Table 1 Comparison of the prevalence of AUID among our center and adult reference centers of other countries
PUMCH:北京协和医院; AUID:自身炎症性疾病;FMF:家族性地中海热;TRAPS:TNF受体相关周期性综合征;CAPS:冷炎素相关周期性综合征;MKD:甲羟戊酸激酶缺乏症/高IgD综合征;NLRP12-AD:NLRP12相关自身炎症性疾病;BS:Blau综合征;PFAPA:周期性发热-阿弗它口炎-咽炎-淋巴结炎综合征;*该研究中大部分成人AUID患者均为NOD2相关自身炎症性疾病(NOD2-associated autoinflammatory disease, NAID)(新近报道的与NOD2基因变异相关的一种AUID)
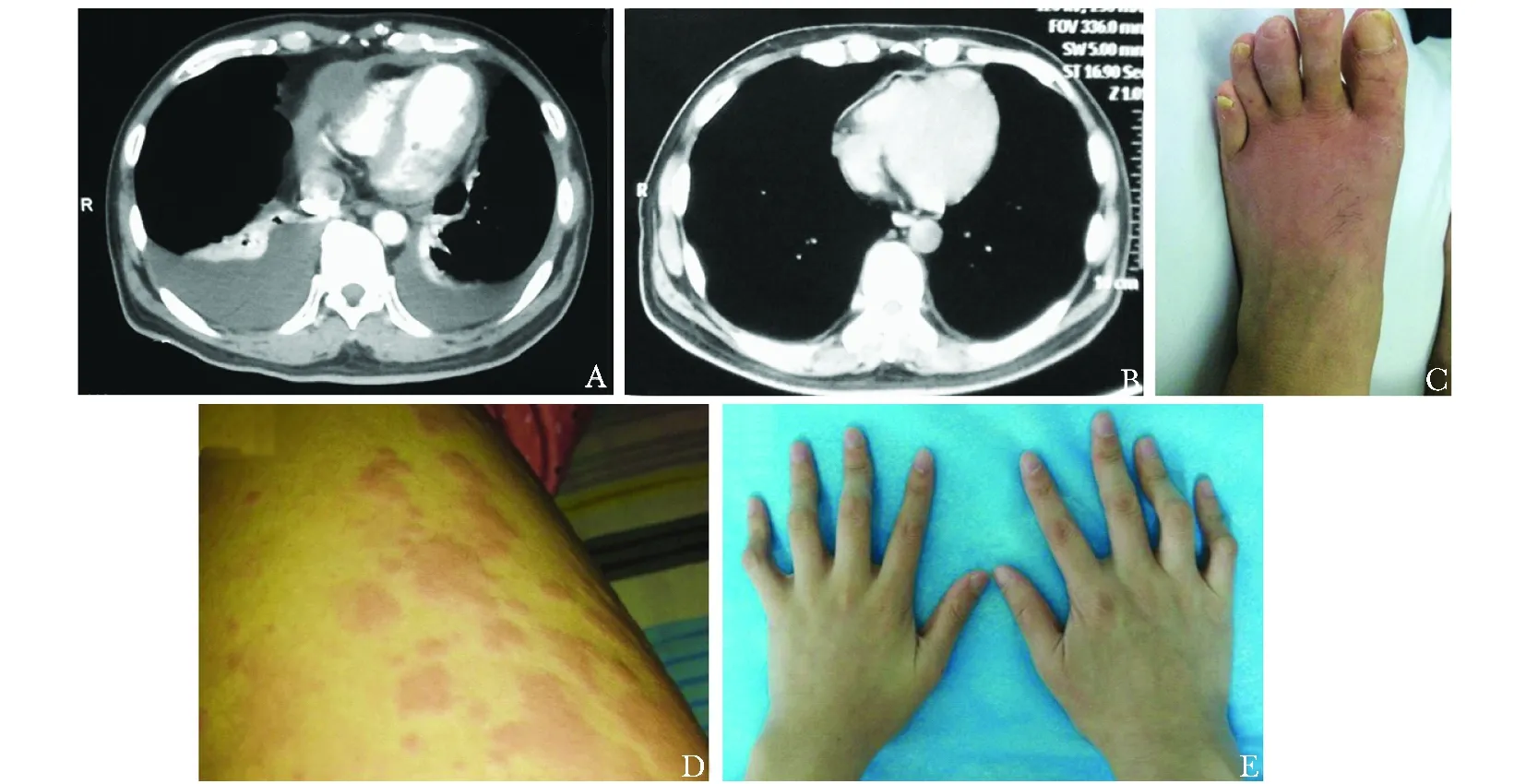
图2成人单基因AUID临床表现
A:家族性地中海热患者胸腔积液,治疗前;B:家族性地中海热患者胸腔积液,治疗后;C:冷炎素相关周期性综合征患者足部片状红斑;D:NLRP12自身炎症性疾病患者肢体荨麻疹;E:Blau综合征患者指挛缩
Fig2Clinical manifestations of adult-onset monogenic AUID
A:Pleural effusion of a patient diagnosed as familial Mediterranean fever before treatment; B:Pleural effusion of a patient diagnosed as familial Mediterranean fever after treatment; C: Erythema on foot of a patient with cryopyrin-associated periodic syndrome; D:Urticarial on limbs of a patient with NLRP12-autoinflammtory disease; E:Camptodactyly in a patient with Blau syndrome; AUID:自身炎症性疾病
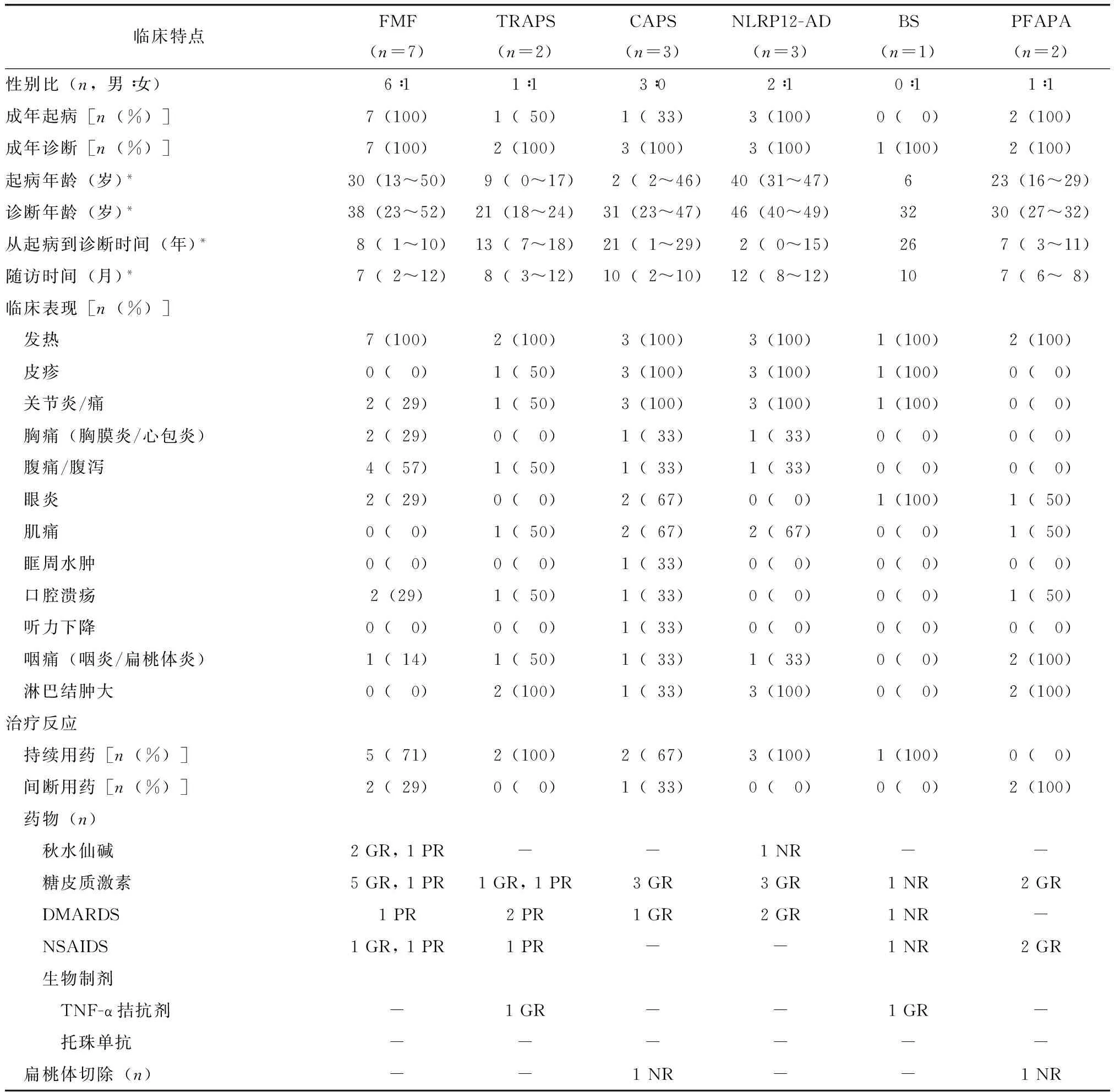
表2 本中心18例成人AUID患者临床表现、治疗和转归Table 2 Summary of the main clinical features, treatment and outcome of the 18 adult patients with AUID in our center
AUID:自身炎症性疾病;*数据以中位数(范围)表示;DMARDS:慢作用抗风湿药;NSAIDS:非甾类抗炎药;FMF:家族性地中海热;TRAPS:TNF受体相关周期性综合征;CAPS:冷炎素相关周期性综合征;NLRP12-AD:NLRP12相关自身炎症性疾病;BS:Blau综合征;PFAPA:周期性发热-阿弗它口炎-咽炎-淋巴结炎综合征;GR:有效;PR:部分有效;NR:无效
本组成人FMF患者均有周期性发热(100%),其次是腹痛/腹泻(57%),1例患者甚至因为腹痛而行阑尾切除术。其他表现包括关节炎/痛、胸痛、结膜炎和口腔溃疡(各29%),皮疹发生率为0。与国外文献报道相比,上述症状发生率相似,而肌痛发生率更低(0vs. 33%)。
本组成人TRAPS患者发热(100%)、皮疹(50%)、关节炎/痛(50%)、肌痛(50%)、淋巴结肿大(100%)以及咽痛(50%)发生率与文献报道相似,而腹痛/腹泻的发生率更高(50%vs. 17%)。
3例CAPS患者的临床表现与国外文献比较,发热(100%vs. 37.5%)、皮疹(100%vs. 0)、关节炎/痛(100%vs. 75%)、淋巴结肿大(33%vs. 12.5%)更常见,而眼炎(67%)、肌痛(67%)、胸痛(33%)、腹痛/腹泻(33%)、眶周水肿(33%)、听力下降(33%)等症状的发生率相似。
3例NLRP12-AD患者与国外文献比较,皮疹(100%vs. 44.4%)、关节炎/痛(100%vs. 33.3%)、肌痛(67%vs. 33.3%)、淋巴结肿大(100%vs. 11.1%)发生率更高,发热(100%)、胸痛(33%)、腹痛/腹泻(33%)、眼炎(0)等表现的发生率相似。
此外,1例BS患者具备非常典型的BS关节炎、皮炎和眼炎三联征,同时有阳性家族史。2例PFAPA综合征患者则全部具备周期性发热、口腔溃疡、咽炎和颈部淋巴结肿大的临床表现。结果均与文献报道相似。
PUMCH中心成人AUID基因型特点
16例成人单基因AUID患者检测出19个基因变异中,明确的、常常与较严重临床表型相关的致病性基因突变仅6个(31.6%),其余13个(68.4%)基因变异均为低外显变异、新发变异或单核苷酸多态性;7例FMF患者均非纯合突变,分别为杂合突变5例(71.4%),复合杂合突变2例(28.6%)(表3)。
讨 论
AUID是一组最近10余年才逐渐被认识的疾病,由于其遗传性特点而常常在幼年时期起病。该组疾病相对罕见,临床表现又常与感染、肿瘤或其他自身免疫性疾病相似,故而诊断十分困难,尤其在成人起病的AUID患者,诊断更显复杂而具有挑战性。在许多儿童AUID研究中,拟诊AUID患者进行基因检测最终能够获得阳性结果的比例不足20%[10,22],而国外成人AUID中心研究报道中基因检测结果阳性率也与之相仿(17%)[10,17]。Cantarini等[9]在2011年曾经提出一个针对成人FMF和TRAPS的诊断评分,依据此临床项目评分,最终获得MEFV和TNFRSF1A基因检测阳性结果的敏感性和特异性高达78%和72%。在此临床诊断评分中,起病年龄、周期性发热的家族史、胸痛、腹痛和皮疹这几项临床表现与基因检测阳性率的相关性更高。本院AUID中心对拟诊AUID患者进行基因检测,阳性率达43.2%,显著高于既往文献报道[10,17,22]。这一方面说明, 本院AUID中心对于成人AUID的认识和诊治具有较高水准。另一方面说明,如果能够很好地应用临床诊断评分,对临床拟诊AUID患者进行全面评估和筛选,最终获得基因检测结果阳性的几率会很大程度提高。
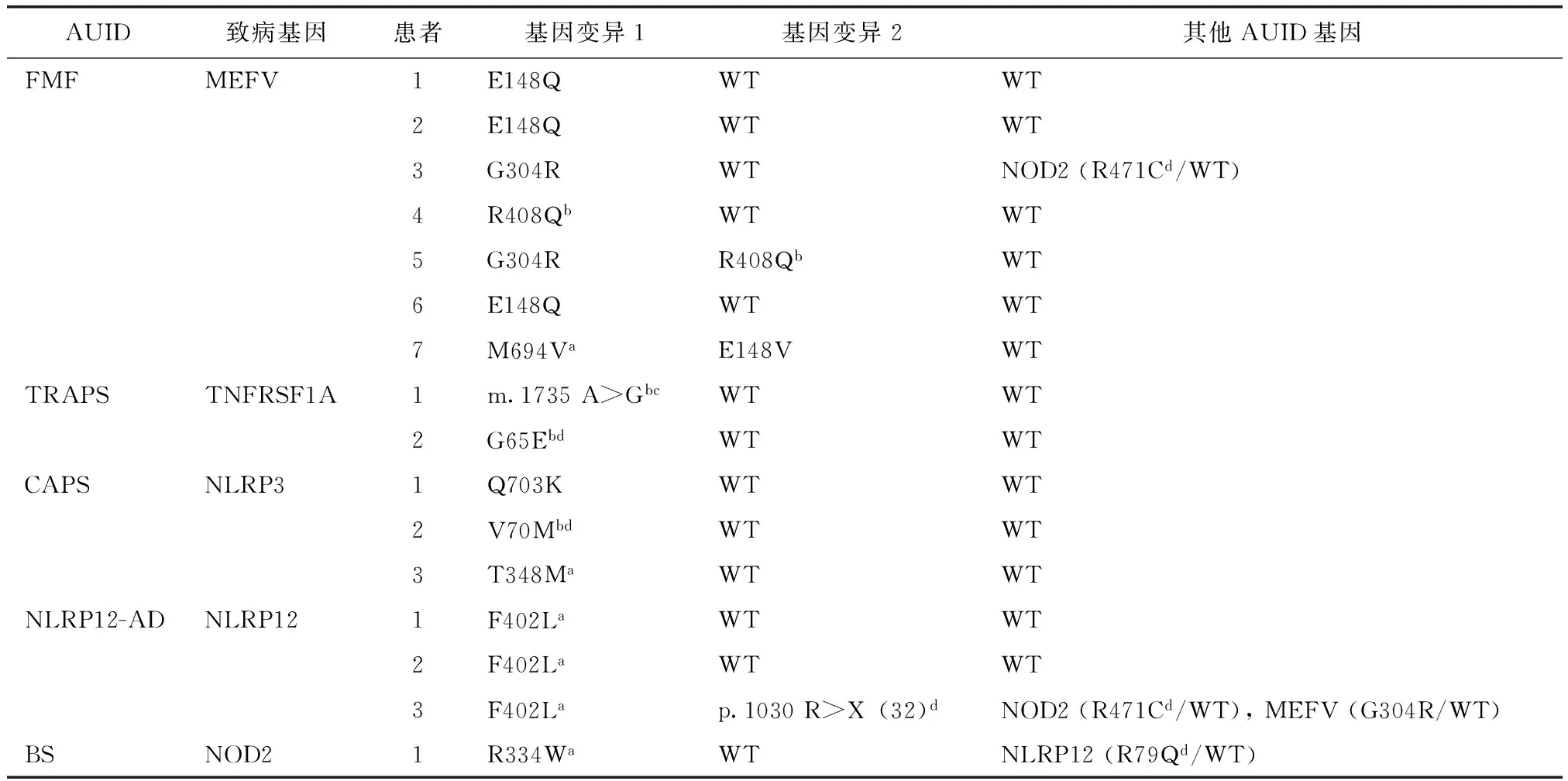
表3 本中心16例单基因成人AUID患者的基因型Table 3 Genotypes of the 16 adult patients with monogenetic AUID in our center
AUID:自身炎症性疾病;FMF:家族性地中海热;TRAPS:TNF受体相关周期性综合征;CAPS:冷炎素相关周期性综合征;NLRP12-AD:NLRP12相关自身炎症性疾病;BS:Blau综合征;WT:野生型;a明确的致病性基因变异;b常见单核苷酸多态性(可能、但并不总是非致病性基因变异),余为低外显率基因变异;c非编码区意义未明的基因变异;d新的基因变异
FMF一直被认为是一种常染色体隐性遗传性疾病,确诊有赖于MEFV基因纯合突变。但是越来越多的研究发现,符合FMF临床诊断标准[23]的患者中有大约20%~30%是MEFV杂合突变[24- 25]。诸多研究表明,MEFV杂合突变结合临床表现仍可以获得较高的诊断敏感性和特异性[25- 26]。而关于FMF的发病年龄,尽管儿童起病多见,但成人起病并非罕见,文献报道18岁以上发病者可达10%~20%,甚至达到30%之多[12,27]。成人起病的FMF被延迟诊断时间甚至长达5~6年[27- 28]。本研究中7例FMF患者均为成年起病,平均发病年龄30岁,其中5例(71.4%)患者20岁以后才发病,而所有患者平均延迟诊断长达8年。提示对于FMF的认识还有待进一步提高。
本研究与国外成人AUID中心的研究结果[17,27,29]均发现,成人FMF患者较幼年起病者关节炎/痛和丹毒样皮疹的发生率更低,临床表现更轻,对治疗反应较好。通过基因型特点分析不难发现,成人FMF患者均为MEFV基因杂合突变或复合杂合突变,而不是纯合突变,并且基因变异多为低外显性,由此使得患者临床症状更轻微或更加不典型[24,30]。本研究还发现,尽管文献报道激素对于治疗FMF无效,但是本组患者较多仍对激素治疗有效或者部分有效,加用秋水仙碱之后效果更佳。提示中国汉族成人FMF存在与高加索人群不同的特点,还需要更多病例、更长期的随诊观察。
TRAPS的平均发病年龄是3岁,但是也有成年后发病的报道,后者常与低外显TNFRSF1A基因变异相关,临床表型更轻,淀粉样变的发生率更低[31]。本研究2例TRAPS患者中1例出生后不久起病,另1例17岁起病。尽管2例患者的基因检测结果是TNFRSF1A基因新发变异或者非编码区意义未明变异,但是结合患者临床表现,以及对传统抗炎药物或TNF-α拮抗剂治疗反应良好,最终诊断TRAPS。笔者认为,尽管这2例患者的基因变异意义未明,仍然可能在TRAPS的发病中起到了作用,其是否为致病性变异尚有待更多病例积累以及进一步转化医学研究证实。
本研究和国外成人AUID中心研究发现一致,CAPS完全可以成年后发病[11,17]。文献已有报道NLRP3基因T348M是致病性基因变异[16],而低外显变异Q703K也是致病性基因变异,可能导致疾病晚发且症状轻微[11,32]。本组3例CAPS患者中,1例T348M变异者2岁发病,而另1例Q703K变异者43岁才发病。3例CAPS患者均诊断为Muckle-Wells综合征,其中2例幼年起病的患者直至20多年后方获得确诊。
除本文3例患者外,其他文献也有成人起病的NLRP12-AD报道[33- 34]。在目前已有报道的NLRP12基因突变中以F402L最常见,本组3例NLRP12-AD患者均为该位点变异。此外,本研究中的1例BS患者尽管6岁就起病,却直至26年后32岁时方获得确诊,此时患者已经出现双目失明、双手关节挛缩等后遗症,并且由于未能获得及时和适宜的遗传咨询,家族中已有多人罹患此病。这更提示我们临床医师应提高对该病的认识和诊治水平。
PFAPA综合征诊断标准包括起病年龄小于5岁[15],而事实上已有许多文献报道成人起病的PFAPA综合征[5,21],平均发病年龄21~26岁。本组2例PFAPA综合征患者均成年起病,平均发病年龄23岁。PFAPA综合征除周期性发热之外,还常常具有阿弗它口炎、咽炎和淋巴结炎三联征或其中两种症状。扁桃体切除对于缓解疾病无效,而采取在发作开始时顿服激素(1次或者2次)的方案可以显著缓解症状。目前尚未发现任何PFAPA综合征致病基因,该病被认为可能是一种多基因AUID。
综上,成人AUID并不少见,患者可能是幼年起病延迟至成年方获得诊断,也完全可以成年后才发病,而对于后者的识别和诊断更加困难。成人AUID以FMF、CAPS和NLRP12-AD相对常见。由于成人AUID患者基因型多为低外显基因变异,临床表现轻微或不典型,易导致诊断的延误。成人拟诊AUID患者的基因检测结果需慎重解读,建议转诊至成人AUID中心专病门诊,由有经验的风湿免疫科医师提供适宜的诊断和治疗。
[1]Hull KM, Drewe E, Aksentijevich I, et al. The TNF receptor-associated periodic syndrome(TRAPS): emerging concepts of an autoinflammatory disorder[J]. Medicine(Baltimore), 2002,81:349- 368.
[2]Theofilopouos AN, Gonzalez-Quintial R, Lawson BR, et al. Sensors of the immune system: their link to rheumatic diseases [J]. Nat Rev Rheumatol, 2010, 6:146- 156.
[3]Doria A, Zen M, Bettio S, et al. Autoinflammation and autoimmunity: Bridging the divide [J]. Autoimmunity Reviews, 2012, 12:22- 30.
[4]Federici S, Caorsi R, Gattorno M. The autoinflammatory diseases[J]. Swiss Med Wkly, 2012, 142:w13602.
[5]Cantarini L, Vitale A, Bartolomei B, et al. Diagnosis of PFAPA syndrome applied to a cohort of 17 adults with unexplained recurrent fevers[J]. Clin Exp Rheumatol, 2012,30:269- 271.
[6]Chandrakasan S, Chiwane S, Adams M, et al. Clinical and genetic profile of children with periodic fever syndromes from a single medical center in South East Michigan[J]. J Clin Immunol, 2014,34:104- 113.
[7]Rigante D. The fresco of autoinfammatory diseases from the pediatric perspective[J]. Autoimmun Rev, 2012,11:348- 356.
[8]Cantarini L, Lucherini OM, Iacoponi F, et al. Development and preliminary validation of a diagnostic score for identifying patients affected with adult-onset autoinflammatory disorders[J]. Int J Immunopathol Pharmacol, 2010,23:1133- 1141.
[9]Cantarini L, Iacoponi F, Lucherini OM, et al. Validation of a diagnostic score for the diagnosis of autoinflammatory diseases in adults[J]. Int J Immunopathol Pharmacol, 2011,24:695- 702.
[10] Muscari I, Iacoponi F, Cantarini L, et al. The diagnostic evaluation of patients with potential adult-onset autoinflammatory disorders: our experience and review of the literature[J]. Autoimmun Rev, 2012,12:10- 13.
[11] Cantarini L, Vitale A, Lucherini OM, et al. Childhood versus adulthood-onset autoinflammatory disorders: myths and truths intertwined[J]. Reumatismo, 2013,65:55- 62.
[12] Cantarini L, Rigante D. Adult-onset autoinfammatory disorders: a still debated entity?[J]. Clin Exp Rheumatol, 2015,33:137- 140.
[13] Cantarini L, Vitale A, Lucherini OM, et al. The labyrinth of autoinflammatory disorders: a snapshot on the activity of a third-level center in Italy[J]. Clin Rheumatol, 2015,34:17- 28.
[14] Kastner DL, Aksentijevich I, Goldbach-Mansky R. Autoinflammatory disease reloaded: a clinical perspective[J]. Cell, 2010, 140:784- 790.
[15] Thomas KT, Feder HM Jr, Lawton AR, et al. Periodic fever syndrome in children[J]. J Pediatr, 1999,135:15- 21.
[16] Shinar Y, Obici L, Aksentijevich I, et al. Guidelines for the genetic diagnosis of hereditary recurrent fevers[J]. Ann Rheum Dis, 2012,71:1599- 1605.
[17] Hernández-Rodríguez J, Ruíz-Ortiz E, Tomé A, et al. Clinical and genetic characterization of the autoinflammatory diseases diagnosed in an adult reference center[J]. Autoimmun Rev, 2016,15:9- 15.
[18] Yao Q, Lacbawan F, Li J. Adult autoinflammatory disease frequency and our diagnostic experience in an adult autoinflammatory clinic[J]. Semin Arthritis Rheum, 2016,45:633- 637.
[19] De Pieri C, Vuch J, Athanasakis E, et al. F402L variant in NLRP12 in subjects with undiagnosed periodic fevers and in healthy controls[J]. Clin Exp Rheumatol, 2014,32:993- 994.
[20] Sfriso P, Caso F, Tognon S, et al. Blau syndrome, clinical and genetic aspects[J]. Autoimmun Rev, 2012,12:44- 51.
[21] Padeh S, Stoffman N, Berkun Y. Periodic fever accompanied by aphthous stomatitis, pharyngitis and cervical adenitis syndrome(PFAPA syndrome) in adults[J]. Isr Med Assoc J, 2008,10:358- 360.
[22] Simon A, van der Meer JW, Vesely R, et al. Approach to genetic analysis in the diagnosis of hereditary autoinflammatory syndromes[J]. Rheumatology(Oxford), 2006,45:269- 273.
[23] Pras M. Familial Mediterranean fever: from the clinical syndrome to the cloning of the pyrin gene[J]. Scand J Rheumatol, 1998, 27:92- 97.
[24] Jéru I, Hentgen V, Cochet E, et al. The risk of familial Mediterranean fever in MEFV heterozygotes: a statistical approach[J]. PloS One, 2013; 8: e68431.
[25] Hentgen V, Grateau G, Stankovic-Stojanovic K, et al. Familial Mediterranean fever in heterozygotes: are we able to accurately diagnose the disease in very young children?[J]. Arthritis Rheum, 2013, 65:1654- 1662.
[26] Ozçakar ZB, Yalçinkaya, Cakar N, et al. Application of the new pediatric criteria and Tel Hashomer criteria in heterozygous patients with clinical features of FMF[J]. Eur J Pediatr, 2011,170:1055- 1057.
[27] Sayarlioglu M, Cefle A, Inanc M, et al. Characteristics of patients with adult-onset familial Mediterranean fever in Turkey: analysis of 401 cases[J]. Int J Clin Pract, 2005, 59: 202- 205.
[28] Nobakht H, Zamani F, Ajdarkosh H, et al. Adult-onset familial Mediterranean fever in northwestern Iran: clinical feature and treatment outcome[J]. Middle East J Dig Dis, 2011,3:50- 55.
[29] 沈敏, 唐琳, 李健, 等. 中国汉族成人起病的家族性地中海热3例及文献复习[J]. 中华临床免疫和变态反应杂志, 2016, 10:75- 79.
[30] Marek-Yagel D, Berkun Y, Padeh S, et al. Clinical disease among patients heterozygous for familial Mediterranean fever[J]. Arthritis Rheum, 2009,60:1862- 1866.
[31] Cantarini L, Rigante D, Meerlini G, et al. The expanding spectrum of low-penetrance TNFRSF1A gene variants in adults presenting with recurrent inflammatory attacks: clinical manifestations and long-term follow-up[J]. Semin Arthritis Rheum, 2014,43:818- 823.
[32] Vitale A, Lucherini OM, Galeazzi M, et al. Long-term clinical course of patients carrying the Q703K mutation in the NLRP3 gene: a case series[J]. Clin Exp Rheumatol, 2012,30:943- 946.
[33] Vitale A, Rigante D, Maggio MC, et al. Rare NLRP12 variants associated with the NLRP12-autoinflammatory disorder phenotype: an Italian case series[J]. Clin Exp Rheumatol, 2013, 31:155- 156.
[34] 唐琳, 侍效春, 李健, 等. NLRP12自身炎症性疾病报道及28例文献[J]. 中华临床免疫和变态反应杂志, 2015, 9:250- 255.
