ARTICLE Structural and Infrared Spectroscopic Study on Solvation of Acetylene by Protonated Water Molecules†
2016-04-08XingtoKongXinLeiQinqinYunBingbingZhngZhiZhoDongYngShukngJingDongxuDiLingJingStteKeyLbortoryofMoleculrRectionDynmicsCollbortiveInnovtionCenterofChemistryforEnergyndMterilsDlinInstituteofChemiclPhysicsChineseAcde
Xing-to Kong,Xin Lei,Qin-qin Yun,Bing-bing Zhng,b,Zhi Zho,c,Dong Yng,Shu-kng Jing,Dong-xu Di,Ling Jing∗.Stte Key Lbortory of Moleculr Rection Dynmics,Collbortive Innovtion Center of Chemistry for Energy nd Mterils,Dlin Institute of Chemicl Physics,Chinese Acdemy of Sciences,Dlin 116023,Chinb.Stte Key Lbortory of Fine Chemicls,Dlin University of Technology,Dlin 116024,Chinc.Key Lbortory of Mterils Modi fi ction by Lser,Ion nd Electron Bems,Dlin University of Technology,Ministry of Eduction,Dlin 116024,Chin
ARTICLE Structural and Infrared Spectroscopic Study on Solvation of Acetylene by Protonated Water Molecules†
Xiang-tao Konga,Xin Leia,Qin-qin Yuana,Bing-bing Zhanga,b,Zhi Zhaoa,c,Dong Yanga,
Shu-kang Jianga,Dong-xu Daia,Ling Jianga∗
a.State Key Laboratory of Molecular Reaction Dynamics,Collaborative Innovation Center of Chemistry for Energy and Materials,Dalian Institute of Chemical Physics,Chinese Academy of Sciences,Dalian 116023,China
b.State Key Laboratory of Fine Chemicals,Dalian University of Technology,Dalian 116024,China
c.Key Laboratory of Materials Modi fi cation by Laser,Ion and Electron Beams,Dalian University of Technology,Ministry of Education,Dalian 116024,China
(Dated:Received on November 19,2015;Accepted on December 11,2015)
The e ff ect of solvation on the conformation of acetylene has been studied by adding one water molecule at a time.Quantum chemical calculations of the H+(C2H2)(H2O)n(n=1−5) clusters indicate that the H2O molecules prefer to form the OH··π interaction rather than the CH··O interaction.This solvation motif is di ff erent from that of neutral(C2H2)(H2O)n(n=1−4)clusters,in which the H2O molecules prefer to form the CH··O and OH··C H-bonds.For the H+(C2H2)(H2O)ncationic clusters,the fi rst solvation shell consists of one ring structure with two OH··π H-bonds and three water molecules,which is completed at n=4.Simulated infrared spectra reveal that vibrational frequencies of OH··π H-bonded O−H stretching a ff ord a sensitive probe for exploring the solvation of acetylene by protonated water molecules.Infrared spectra of the H+(C2H2)(H2O)n(n=1−5)clusters could be readily measured by the infrared photodissociation technique and thus provide useful information for the understanding of solvation processes.
Key words:Acetylene,Water,Solvation,Infrared photodissociation spectroscopy,Quantum chemical calculation
†Part of the special issue for“the Chinese Chemical Society’s 14th National Chemical Dynamics Symposium”.
∗Author to whom correspondence should be addressed.E-mail: ljiang@dicp.ac.cn.
I.INTRODUCTION
Hydrogen-bonded interactions are of considerable interest because of their signi fi cant importance in physical,chemical,atmospheric,biological sciences,and so on[1−7].The classical hydrogen bonds(H-bonds)are usually de fi ned as A−H··B interactions,where A−H is a proton donating bond and B is a proton accepting center that has at least one lone electron pair. A and B are electronegative atoms,such as O,N, F,and Cl.Extensive e ff orts have also been made to study new types of H-bonds,which include nonconventional proton donors(i.e.,C−H)and proton acceptors (i.e.,π-bonded functional groups),as well as dihydrogen bonds[8−13].
Fully rotationally resolved spectra have demonstrated that water is positioned above the benzene plane,forming the OH··π H-bonded interactions[14]. Resonantion-dipinfraredspectroscopyofthe C6H6(H2O)n(n=1−7)clusters has indicated that there exist both classical OH··O and nonconventional OH··π H-bonded interactions[15].The NH··π H-bonded interactions have been detected in the ammonia-benzene dimer by high-resolution optical and microwave spectra [16].High-level ab initio calculations have exhibited that the OH··π H-bond in C6H6-H2O is stronger than the NH··π H-bond in C6H6-NH3,and their directionality is mainly controlled by the electrostatic interaction [17].The CH··π H-bonded energy in C6H6-CH4is determined to be 4.31−4.73 kJ/mol by mass analyzed threshold ionization spectroscopy,which is consistent with the theoretical value calculated by the CCSD(T) method[18].In the interactions of ethylene with the fi rst-row hydrides(CH4,NH3,H2O,and HF),π H-bonds become stronger from CH4to HF,which is highly correlated to inductive energy[19].
The OH··π H-bond could also be formed in the interactions of water with the simplest alkyne,acetylene. Previous studies have revealed that(C2H2)(H2O)has two con fi gurations,in which either H2O acts as proton acceptor to form CH··O H-bond or C2H2serves as proton acceptor to form OH··π H-bond.The CH··O H-bond is stronger than the OH··π H-bond,and the barrier for the isomerization from the latter to the former is very low(0.75 kJ/mol),suggesting that the interconversion readily occurs[11,20].Theoretical inves-tigations of neutral(C2H2)(H2O)n(n=1−4)clusters have indicated that the interactions between acetylene and water are mainly composed by CH··O and OH··C H-bonds[21,22].When acetylene interacts with protonated water molecule(H3O+),only one stable con fi guration with OH··π H-bond is formed,which binding energy was predicted to be 81.13 kJ/mol at MP2/6-311++G(3df,3pd)level[5].So far,much less work has been done for the systematic study on the solvation of C2H2by protonated water clusters,leaving that the issues how the excess proton a ff ects solvation motif of C2H2as compared to the neutral water and how the OH··π H-bond varies with sequential hydration remain open.
Herein,we present a study on the solvation of C2H2by a series of protonated water clusters.Electronic structure calculations of the H+(C2H2)(CO2)n(n=1−5) clusters reveal that the H2O molecules prefer to form the OH··π interaction rather than the CH··O interaction.The fi rst solvation shell consists of one ring structure with two OH··π H-bonds and three water molecules,which is completed at n=4.Simulated IR spectra exhibit that vibrational frequencies of OH··π H-bonded O−H stretching a ff ord a sensitive probe for exploring the solvation of acetylene by adding one water molecule at a time.
II.THEORETICAL METHODS
Quantum chemical calculations are carried out using Gaussian 09 program suite[23].Initial con fi gurations are built on the basis of the relevant structures reported in Refs.[5,21,22].Previous investigations have demonstrated that the structures and energetics of H-bonded complexes could be properly predicted by the M06-2X hybrid functional[24−27],which functional is employed for the present calculations.The aug-ccpVDZ basis set is used for C,H,and O atoms.Tight convergence of the optimization and the self-consistent fi eld procedures is imposed,and an ultra fi ne grid is utilized.Relative energies include zero-point vibrational energies.Harmonic vibrational frequencies are calculated at the same level.All reported structures are true minima without imaginary vibrational frequencies. Simulated IR spectra are derived from M06-2X/aug-ccpVDZ harmonic vibrational frequencies and intensities. Harmonic vibrational frequencies are scaled by a factor of 0.933,which is determined by the comparison of simulated vibrational frequencies of the bridged proton stretch in the nonclassical H+(C2H2)ion with experimental value[28].IR stick spectra are convoluted by a Gaussian line shape function with a width of 10 cm−1. The quantum theory of“atoms in molecules”calculations are performed by the Multiwfn program at the M06-2X/aug-cc-pVDZ level[29].
III.RESULTS AND DISCUSSION
A.Solvation motifs and IR spectra
Several representative low-lying structures of the H+(C2H2)(H2O)n(n=1−5)clusters are presented in Fig.1.The structures are labeled according to the number of H2O molecules and relative energies.For each cluster up to n=5,simulated IR spectra of the representative low-lying isomers are shown in Figs.2−5. Scaled harmonic vibrational frequencies and intensities of free O−H stretching,OH··π H-bonded O−H stretching,OH··O H-bonded O−H stretching,and CC2H2in the lowest-lying isomers of (n=1−5)are listed in Table I.
1.n=1
As illustrated in Fig.1,one OH group of the H3O+moiety in the isomer 1A forms one OH··π H-bond with π electrons of C2H2,leaving other two OH groups free.
Four kinds of absorption peaks are mainly observed in 1A(Fig.2).The frequencies at 3536 and 3449 cm−1 are assigned to the free O−H stretching vibration of H3O+(labeled free νOH)(Table I).The frequency at 3134 cm−1corresponds to the C−H stretching vibrations of C2H2(labeled νCH).Sharp peak at 2036 cm−1 belongs to the OH··π H-bonded O−H stretching(labeled νOH···π).The C≡C stretching vibration(labeled νC≡C)is predicted at 1920 cm−1.
2.n=2
On the basis of 1A,the second H2O either binds to H3O+(isomer 2A)or one CH end of acetylene (isomer 2B).The isomer 2B consists of one OH··π H-bond and one CH··O H-bond,which lies 64.03 kJ/mol above 2A.
In the simulated IR spectrum of 2A(Fig.2),the free νOHmodes appear at 3650 and 3556 cm−1.The frequency at 3158 cm−1corresponds to the νCHmotion. The peak at 2813 cm−1is attributed to the OH··π H-bonded O−H stretching,which is remarkably blueshifted by 777 cm−1with respect to that in 1A.A new type of absorption peak is observed around 1963 cm−1 in 2A as compared to 1A,which is assigned to the OH··O H-bonded O−H stretching(labeled νOH···O). For 2B,the free νOH,νCH,and νOH···πmodes are calculated around 3560,3000,and 1800 cm−1,respectively.
3.n=3
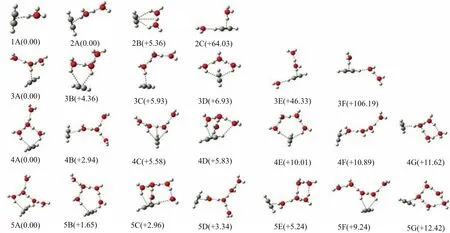
FIG.1 Optimized structures of the H+(C2H2)(H2O)n(n=1−5)clusters(C:gray;H:light gray;O:red).Relative energies are given in parenthesis with unit of kJ/mol.

TABLE I Scaled vibrational frequencies(in cm−1)and intensities(km/mol,in parenthesis)of free O−H stretching(free νOH),OH··O H-bonded O−H stretching(νOH···O),OH··π H-bonded O−H stretching(νOH···π),and C−H stretching of C2H2(νCH)for the lowest-lying isomers of H+(C2H2)(H2O)n(n=1−5).
The lowest energy isomer(3A)could be regarded as the derivative of 2A,in which the third H2O occupies the remaining free OH site of H3O+(Fig.1).In the next energetically low-lying isomer 3B(+4.36 kJ/mol), one H-bonded ring is formed by two OH··π H-bonds and one OH··O H-bond.3C(+5.93 kJ/mol)is evolved from 2A,in which the third water is attached by one H-bonded interaction with the second water.In 3D (+6.93 kJ/mol),two terminal water molecules in the H7O3+chain form two OH···π H-bonds with C2H2,resulting in one H-bonded three-water ring.The isomers 3E(+46.33 kJ/mol)and 3F(+106.19 kJ/mol)could be viewed as the derivative of 2C,in which the third water binds to the OH group of H3O+and H2O,respectively.

FIG.2 Simulated IR spectra of the optimized isomers of H+(C2H2)(H2O)1,2.Assignments of free O−H stretching, OH··π H-bonded O−H stretching,OH··O H-bonded O−H stretching,and C−H stretching of C2H2,are indicated in green,red,magenta,and blue,respectively.
In the simulated IR spectra of 3A−3F(Fig.3),the free O−H stretching vibrations weakly appear around 3600 cm−1.The intensities of C−H stretching vibrations are very weak(around 3200 cm−1)in 3A−3D,but recover remarkably in 3E and 3F because of the formation of CH··O H-bonds.The νOH···πmotion presents at 3078,3423,3247,3524/3518,2655,and 1725 cm−1 in 3A−3F,respectively.The νOH···Opeaks are observed around 1800−2500 cm−1in 3A−3E and 3400 cm−1in 3F.
4.n=4 and 5
For the n=4 cluster,the isomer 4A could be viewed as the derivative of 3D,in which the fourth water binds to the free OH group of the H3O+moiety(Fig.1).4B is the analogy of 3C,which lies 2.94 kJ/mol higher in energy above 4A.In 4C(+5.58 kJ/mol),the fourth water is attached to the free OH group of the H5O2+moiety of 3B,of which the shared proton also forms one H··π interaction with acetylene with the bond distance of 2.97˚A[30].Analogous to 4A,4D holds one additional CH··O H-bond between H2O and C2H2,which lies+5.83 kJ/mol above 4A.In the structure of 4E (+10.01 kJ/mol),one H-bonded ring is formed by two OH··π H-bonds and four H-bonds.In 4E,the distance between the shared proton and the center of C2H2is so long(4.41˚A)that there is no H··π interaction.In 4F(+10.89 kJ/mol),one terminal of the H9O4+chain forms OH··π H-bond with C2H2,similar to 3C.In 4G(+11.62 kJ/mol),one ring containing four water molecules is generated and the H3O+moiety is incorporated into the formation of one OH··π H-bond.For the n=5 cluster,the solvated structures are similar to those of n=4,suggesting that the formation of two OH··π H-bonds with the ring containing three or four water molecules is favorable.

FIG.3 Simulated IR spectra of the six optimized isomers of H+(C2H2)(H2O)3.Assignments of free O−H stretching, OH··π H-bonded O−H stretching,OH··O H-bonded O−H stretching,and C−H stretching of C2H2are indicated in green,red,magenta,and blue,respectively.
In the simulated IR spectra of the n=4 cluster(Fig.4), the νOH···πmotion is calculated at 3539/3532,3355, 3584/3526,3556/3553,3546/3513,3350,and 3117 cm−1 in 4A−4G,respectively.The νOH···Omodes are sharply observed in all the isomers,the positions are distinctively di ff erent throughout the seven isomers.Interestingly,the characteristic vibrations of H5O2+Zundel ion present around 1600 and 1080 cm−1in 4C and 4E,respectively[31].
As depicted in Fig.5,the νOH···πmotion is predicted to be 3583/3544,3564/3512,3562/3557,3401,3402, 3613/3516,and 3223 cm−1in 5A−5G,respectively. Several peaks of νOH···Omodes remarkably appear in all the isomers.However,the vibrations of H5O2+Zundel is absent from the n=5 cluster.
B.General trend
It can be seen from the aforementioned solvation motifs that the H2O molecules in the n=1−5 clusters prefer to form the OH··π interaction rather than the CH··O interaction.The fi rst solvation shell consists of one ring structure with two OH··π H-bonds and three water molecules,which is completed at n=4.Previous studies on the neutral(C2H2)(H2O)n(n=1−4)clusters revealed that the H2O molecules prefer to form the CH··O and OH··C H-bonds with C2H2,and C2H2is involved in the formation of a H-bonded ring starting at n=2.At n=4,the neutral(C2H2)(H2O)4cluster holds a water tetramer interacting with acetylene[21],which is di ff erent from the protonoated H+(C2H2)(H2O)4cluster that contains a water trimer interacting with acetylene.
IR spectra ofthe lowest-lying isomers for the H+(C2H2)(H2O)n(n=1−5)clusters are compared in Fig.6.It can be seen that the intensities of free O−H stretching and C−H stretching are very weak,and their positions slightly change with the increase of cluster size.The OH··π H-bonded O−H stretching is predicted at 2036,2813,3078,3532/3539,and 3544/3583 cm−1for 1A−5A(Table II),respectively,exhibiting an obvious increase with the increase of cluster size.This implies that the OH··π interaction strength is weakened gradually as the number of the water molecule increases,consequently,resulting in a decrease trend of the red-shift(∆νOH···π,Table II)from the antisymmetric stretching vibrational frequency(3756 cm−1)of the free water molecule[32].Furthermore,the∆νOH···πvalues are very similar at n=4 or 5,suggesting that the solvation of acetylene by prontonated water approaches to be converged around n=4 or 5.The νOH···Omotions are also blue-shifted from 2A to 5A,but are split into several peaks and expanded in a broad region at larger clusters,indicating a less sensitive probe than the νOH···πmotion for exploring early stage solvation of acetylene by adding one water molecule at a time.

TABLE II Electron density(ρ(rb)),Laplacian of electron density(■2ρ(rb)),and energy density(HBCP)at the OH··π H-bond critical points in the lowest-lying isomers(1A−? 5A).Scaled vibrational frequencies of OH··π H-bonded O−H stretching(νOH···π)and their red-shifts(∆νOH···π)from the antisymmetric stretching vibrational frequency(3756 cm−1)of the free water molecule are also given.

FIG.4 Simulated IR spectra of the seven optimized isomers of H+(C2H2)(H2O)4.Assignments of free O−H stretching, OH··π H-bonded O−H stretching,OH··O H-bonded O−H stretching,C−H stretching of C2H2,and diagnostic vibration of H5O2+are indicated in green,red,magenta,blue, and orange,respectively.

FIG.5 Simulated IR spectra of the seven optimized isomers of H+(C2H2)(H2O)5.Assignments of free O−H stretching, OH··π H-bonded O−H stretching,OH··O H-bonded O−H stretching,and C−H stretching of C2H2are indicated in green,red,magenta,and blue,respectively.
Topological parameters of OH··π H-bonds at bond critical points are calculated to assess the e ff ect of protonated water molecules on the νOH···πfrequency shift. Electron density(ρ(rb)),Laplacian of electron density(■2ρ(rb)),and energy density(HBCP)of OH··π H-bond at bond critical points(BCPs)in the lowestlying isomers(1A−5A)are summarized in Table II. The ρ(rb)value for 1A−5A is calculated to be 0.0584, 0.0287,0.0362,0.0126/0.0125,and 0.0124/0.0118(Table II),respectively,indicating a monotonic decrease of OH··π H-bond strength.This supports the red-shifts of OH··π H-bonded O−H stretching vibrational frequencies from the free water molecule as a function of the number of water molecule(Fig.7).The negative HBCPvalues for 1A(−0.0134)and 2A(−0.0007)suggest the OH··π H-bonds could be regarded as partially covalent interactions.The HBCPvalues become positive in the 3A−5A clusters,indicating that the OH··π H-bonds could be mainly dominant by electrostatic interactions and thus get weaker as the cluster size increases[5].

FIG.6 Simulated IR spectra of the lowest-lying isomers of H+(C2H2)(H2O)n(n=1−5).Assignments of free O-H stretching,OH··π H-bonded O−H stretching,OH··O H-bonded O−H stretching,and C−H stretching of C2H2are indicated in green,red,magenta,and blue,respectively.
Infrared photodissociation(IRPD)spectroscopy of mass-selected complexes has emerged as a powerful tool for the structural characterization of the gas-phase species[33−36].Under readily achievable experimental conditions,absorption of single IR photon or multiple IR photons by a cluster can induce a measurable increase in the sequence,resulting in IRPD spectra that closely resemble linear absorption spectra.Compared with the conventional vibrational spectroscopy, IRPD has advantages of high selectivity,high sensitivity and being a background-free consequence technique.Considering that the free O−H stretching,H-bonded O−H stretching,and diagnostic vibration of H5O2+have been successfully resolved in the IRPD spectra of a series of mass-selected clusters radiated by optical parametric oscillator/optical parametric ampli fi er table-top laser system or infrared free electron laser source[31,37−39],the predicted IR spectra for the H+(C2H2)(H2O)n(n=1−5)clusters could be readily measured by the IRPD technique and thus a ff ord useful information for the understanding of early stage solvation of acetylene by adding one water molecule at a time.
IV.CONCLUSION
The solvation of acetylene by protonated water molecules has been studied via a cluster model.Quantum chemical calculations of the H+(C2H2)(H2O)n(n=1−5)clusters indicate that the H2O molecules prefer to form the OH··π interaction rather than the CH··O interaction.The fi rst solvation shell consists of one ring structure with two OH··π H-bonds and three water molecules,which is completed at n=4.Simulated IR spectra reveal that vibrational frequencies of OH··π H-bonded O−H stretching a ff ord a sensitive probe for exploring the solvation of acetylene by protonated water molecules.The combination of IRPD technique and theoretical modeling thus provide a vivid physical picture about how protonated water molecules solvate the acetylene.

FIG.7 Red-shifts(∆νOH···π)of OH··π H-bonded O−H stretching vibrational frequencies from the antisymmetric stretching vibrational frequency(3756 cm−1)of the free water molecule and electron density(ρ(rb))of the OH··π H-bond critical points as a function of the number of water molecule(n).
V.ACKNOWLEDGMENTS
This work was supported by the National Natural Science Foundation of China(No.21273232 and No.21327901)and the Key Research Program of the Chinese Academy of Science(No.KGZD-EW-T05). Ling Jiang acknowledges Hundred Talents Program of Chinese Academy of Sciences and Collaborative Innovation Center of Chemistry for Energy and Materials.
[1]G.A.Je ff rey and W.Saenger,Hydrogen Bonding in Biological Structures,Berlin:Springer-Verlag(1991).
[2]G.A.Je ff rey,An Introduction to Hydrogen Bonding, New York:Oxford University Press(1997).
[3]G.R.Desiraju and T.Steiner,The Weak Hydrogen Bond in Structural Chemistry and Biology,New York:Oxford University Press Inc.(1999).
[4]T.Steiner,Angew.Chem.Int.Ed.41,48(2002).
[5]S.Janusz Grabowski,Chem.Rev.111,2597(2011).
[6]N.Heine and K.R.Asmis,Int.Rev.Phys.Chem.34, 1(2015).
[7]P.A.Hunt,C.R.Ashworth,and R.P.Matthews, Chem.Soc.Rev.44,1257(2015).
[8]I.Alkorta,I.Rozas,and J.Elguero,Chem.Soc.Rev. 27,163(1998).
[9]P.Hobza and Z.Havlas,Chem.Rev.100,4253(2000).
[10]S.J.Grabowski,J.Phys.Chem.A 105,10739(2001). [11]L.Sobczyk,S.J.Grabowski,and T.M.Krygowski, Chem.Rev.105,3513(2005).
[12]S.Tsuzuki and A.Fujii,Phys.Chem.Chem.Phys.10, 2584(2008).
[13]B.G.de Oliveira,Phys.Chem.Chem.Phys.15,37 (2013).
[14]S.Suzuki,P.G.Green,R.E.Bumgarner,S.Dasgupta, W.A.Goddard,and G.A.Blake,Science 257,942 (1992).
[15]R.N.Pribble and T.S.Zwier,Science 265,75(1994).
[16]D.A.Rodham,S.Suzuki,R.D.Suenram,F.J.Lovas, S.Dasgupta,W.A.Goddard,and G.A.Blake,Nature 362,735(1993).
[17]S.Tsuzuki,K.Honda,T.Uchimaru,M.Mikami,and K.Tanabe,J.Am.Chem.Soc.122,11450(2000).
[18]K.Shibasaki,A.Fujii,N.Mikami and S.Tsuzuki,J. Phys.Chem.A 110,4397(2006).
[19]P.Tarakeshwar,H.S.Choi,and K.S.Kim,J.Am. Chem.Soc.123,3323(2001).
[20]D.Tzeli,A.Mavridis,and S.S.Xantheas,J.Chem. Phys.112,6178(2000).
[21]D.Tzeli,A.Mavridis,and S.S.Xantheas,Chem.Phys. Lett.340,538(2001).
[22]D.Tzeli,A.Mavridis,and S.S.Xantheas,J.Phys. Chem.A 106,11327(2002).
[23]M.J.Frisch,G.W.Trucks,H.B.Schlegel,G.E. Scuseria,M.A.Robb,J.R.Cheeseman,G.Scalmani, V.Barone,B.Mennucci,G.A.Petersson,H.Nakatsuji,M.Caricato,X.Li,H.P.Hratchian,A.F.Izmaylov,J.Bloino,G.Zheng,J.L.Sonnenberg,M. Hada,M.Ehara,K.Toyota,R.Fukuda,J.Hasegawa, M.Ishida,T.Nakajima,Y.Honda,O.Kitao,H.Nakai, T.Vreven,J.A.Montgomery Jr.,J.E.Peralta,F. Ogliaro,M.J.Bearpark,J.Heyd,E.N.Brothers,K.N. Kudin,V.N.Staroverov,R.Kobayashi,J.Normand,K. Raghavachari,A.P.Rendell,J.C.Burant,S.S.Iyengar,J.Tomasi,M.Cossi,N.Rega,N.J.Millam,M. Klene,J.E.Knox,J.B.Cross,V.Bakken,C.Adamo, J.Jaramillo,R.Gomperts,R.E.Stratmann,O.Yazyev, A.J.Austin,R.Cammi,C.Pomelli,J.W.Ochterski, R.L.Martin,K.Morokuma,V.G.Zakrzewski,G.A. Voth,P.Salvador,J.J.Dannenberg,S.Dapprich,A. D.Daniels,¨O.Farkas,J.B.Foresman,J.V.Ortiz,J. Cioslowski,and D.J.Fox,Gaussian 09,Rev A.02, Wallingford CT:Gaussian,Inc.,(2009).
[24]K.L.Copeland and G.S.Tschumper,J.Chem.Theory Comput.8,1646(2012).
[25]S.R.Gadre,S.D.Yeole,and N.Sahu,Chem.Rev.114, 12132(2014).
[26]X.B.Wang and S.R.Kass,J.Am.Chem.Soc.136, 17332(2014).
[27]Z.Zhao,X.T.Kong,X.Lei,B.B.Zhang,J.J.Zhao, and L.Jiang,Chin.J.Chem.Phys.28,501(2015).
[28]G.E.Douberly,A.M.Ricks,B.W.Ticknor,W.C. McKee,P.v.R.Schleyer,and M.A.Duncan,J.Phys. Chem.A 112,1897(2008).
[29]T.Lu and F.Chen,J.Comput.Chem.33,580(2012).
[
30]S.J.Grabowski,J.Phys.Chem.A 111,13537(2007).
[31]K.R.Asmis,N.L.Pivonka,G.Santambrogio,M. Brummer,C.Kaposta,D.M.Neumark,and L.Woste, Science 299,1375(2003).
[32]L.Jiang,T.Wende,R.Bergmann,G.Meijer,and K. R.Asmis,J.Am.Chem.Soc.132,7398(2010).
[33]E.J.Bieske and O.Dopfer,Chem.Rev.100,3963 (2000).
[34]M.A.Duncan,Int.Rev.Phys.Chem.22,407(2003).
[35]K.R.Asmis and D.M.Neumark,Acc.Chem.Res.45, 43(2012).
[36]A.B.Wolk,C.M.Leavitt,E.Garand,and M.A.Johnson,Acc.Chem.Res.47,202(2014).
[37]J.M.Headrick,E.G.Diken,R.S.Walters,N.I.Hammer,R.A.Christie,J.Cui,E.M.Myshakin,M.A. Duncan,M.A.Johnson,and K.D.Jordan,Science 308,1765(2005).
[38]A.Fujii and K.Mizuse,Int.Rev.Phys.Chem.32,266 (2013).
[39]J.A.Fournier,C.J.Johnson,C.T.Wolke,G.H.Weddle,A.B.Wolk,and M.A.Johnson,Science 344,1009 (2014).
杂志排行

CHINESE JOURNAL OF CHEMICAL PHYSICS的其它文章
- ARTICLE E ffi cient Separation of Ar and Kr from Environmental Samples for Trace Radioactive Noble Gas Detection†
- REVIEW Polarization Dependent Time-Resolved Infrared Spectroscopy and Its Applications†
- ARTICLE Reactions of Group V Metal Atoms with Hydrogen Sul fi de:Argon Matrix Infrared Spectra and Theoretical Calculations†
- ARTICLE Structural Dynamics of Phenyl Azide in Light-Absorbing Excited States: Resonance Raman and Quantum Mechanical Calculation Study†
- ARTICLE Excited-State Proton Transfer and Decay in Hydrogen-Bonded Oxazole System:MS-CASPT2//CASSCF Study†
- ARTICLE Infrared Photodisssociation Spectroscopy of Boron Carbonyl Cation Complexes†