在α-蒎烯生物环氧化反应过程中酯对反应的影响及机理探讨
2016-03-07熊阳覃益民唐爱星韦荟琳刘幽燕广西大学化学化工学院广西南宁530004广西生物炼制重点实验室广西南宁530003
熊阳,覃益民,唐爱星,韦荟琳,刘幽燕,(广西大学化学化工学院,广西 南宁 530004;广西生物炼制重点实验室,广西 南宁 530003)
在α-蒎烯生物环氧化反应过程中酯对反应的影响及机理探讨
熊阳1,覃益民1,唐爱星1,韦荟琳1,刘幽燕1,2
(1广西大学化学化工学院,广西 南宁 530004;2广西生物炼制重点实验室,广西 南宁 530003)
摘要:在以过氧化脲(UHP)为氧源、Nov435为催化剂的体系中进行α-蒎烯生物环氧化反应的研究,重点考察不同酯作为酰基供体对反应的影响。从反应速率和酶批次稳定性综合考虑,以丙酸乙酯为最佳溶剂,30℃下α-蒎烯3h的环氧转化率可达到87%左右,经6批次反应后仍可达到47.6%,发现过酸和H2O2的协同作用对酶稳定性的影响要高于单一因素。酶催化乙酸乙酯、丙酸乙酯、乙酸戊酯、己酸乙酯等不同酰基供体过水解反应均可在60min达到平衡,但过酸平衡浓度和α-蒎烯化学环氧化反应速率受酯影响较大,进而对总反应化学-酶法联合催化环氧化产生影响。最后探讨了丙酸乙酯为唯一酰基供体时的反应机制,认为酶利用丙酸产过酸的反应速率和过酸平衡浓度远低于酯;丙酸乙酯的水解和过水解反应为一对竞争性反应,因此有机体系更有利于环氧化反应。
关键词:丙酸乙酯;α-蒎烯;环氧化反应;失活;脂肪酶;过水解;生物催化
第一作者:熊阳(1989—),男,硕士研究生。E-mail yiyuan35@aliyun. com。联系人:覃益民,博士,教授。E-mail qym6289@sina.com。
1990年BJORKLING等[1]发现多种脂肪酶属(EC 3.1.1.3,三酰基甘油酰基水解酶)具有催化“过水解”(perhydrolysis)反应的功能,即以有机酸和过氧化氢为底物催化合成过氧酸,这是脂肪酶催化的多功能现象[2-3]。化学-酶法联合催化环氧化(chemo-enzymatic epoxidation)是此反应的一个典型应用。如图1所示,过水解反应与环氧化反应耦合,使得过水解反应生成的过氧酸自动将活性氧原子传递至碳碳双键生成环氧化物。与化学催化剂[4]相比,生物法具有安全、绿色工艺以及收率高等优点[5-6]。目前人们已经利用反应途径开展了植物油、烯烃、氯代烃等底物[6-11]的环氧化,Novozym435是此反应中最有效的催化剂。
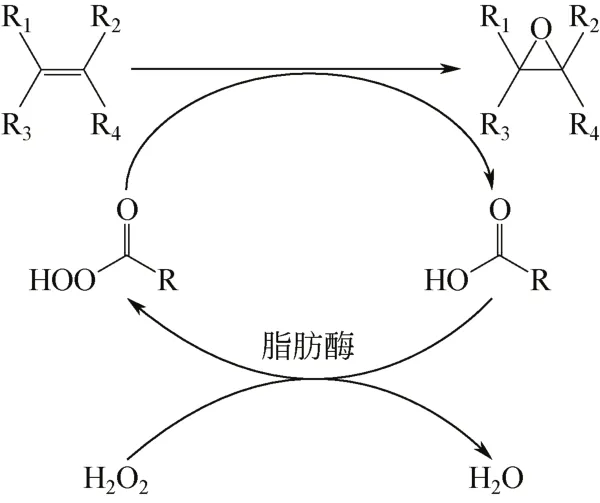
图1 以脂肪酸为酰基供体时酶催化烯烃环氧化原理图[1]
α-蒎烯是自然界中存在量最多的单萜烯烃之一[12],在樟脑、冰片等化学中间体及各种合成香料、生物活性物质制备中起着不可替代的作用。环氧化反应是其深加工的重要途径之一,所获的产物不仅本身有清凉气息、可作为香料,还可进一步合成两百多种香料或有生理活性的精细化学品[13],所以条件温和、选择性高、副反应少的生物法更有利于此类对品质要求较高的医药和香料等功能精细化学品的制备。
在化学-酶法联合催化环氧化反应中,最初的酰基供体多以脂肪酸为酰基供体,在两相体系中进行α-蒎烯环氧化[14-17],虽然底物转化完全,但由于α-蒎烯环氧化物为“酸敏感”物质[18],最高收率仅达到65%,酶也只能使用4批次。因此从反应稳定性、收率等角度考虑,一些研究人员选用碳酸二甲酯等酯类作为酰基供体时,α-蒎烯收率可分别达到85%[19];而XU等[10]以ε-己内酯为溶剂可以实现在两相体系中93%的收率。当以过氧化脲(能缓慢释放H2O2)为氧源时,采用乙酸乙酯为酰基供体和溶剂,反应可获得96%的转化率,最高收率达95%,酶能连续使用7批次,但是酶用量较高(33mg/mL)[11]。
这些研究表明,酰基供体对α-蒎烯环氧化反应有显著影响。首先不同酯的物性会影响H2O2在两相间的分布,而H2O2既是反应物,又是酶的失活剂,其浓度是反应速率和稳定性的关键因素;其次,酯作为过水解反应底物之一,不同酯会生成不同碳链长度过酸,从而可能影响过水解反应速率和α-蒎烯环氧化反应速率。一些研究者发现环氧化体系中存在着中间产物——过酸[20-21],而不同碳链长度的过酸对酶活的影响不同,如TORNVALL等[8]曾将酶置于0.1mol/L过十六烷酸16h后酶活仍保持99.7%,但他们也认为一些短链过酸如过氧乙酸对酶的毒害作用很大。
对于生物催化过程来说,要想实现工业化应用,需要综合考虑反应速率、酶稳定性及产物收率。为此本文选择以过氧化脲为氧源的有机溶剂体系为反应体系,从酶稳定性和转化率两个方面来考察酯种类对过水解反应以及α-蒎烯环氧化反应的影响,并初步探讨了以丙酸乙酯为酰基供体的环氧化反应机理,从而初步解释了丙酸乙酯体系反应活性较高的原因。
1 实验部分
1.1不同酯类对反应的影响
分别以乙酸乙酯、乙酸甲酯、乙酸丁酯、丙酸乙酯、三乙酸甘油酯、乙酸戊酯、己酸乙酯溶液为溶剂,配制α-蒎烯浓度为0.67mol/L的底物溶液。取出1.5mL底物溶液于5mL玻璃瓶中并加入过氧化脲(1.1mmoL),置于平行合成仪(智城公司,型号ZHWY-113H4F)中预热至30℃后加入50mg Novozym435(固定化南极假丝酵母脂肪酶B,购买于美国Sigma–Aldrich)启动反应,250r/min下反应3h后取样气相分析。在丙酸乙酯体系与乙酸乙酯体系中,每个批次反应3h后取样气相分析,脂肪酶用乙腈-水(V/V=9/1)洗去脲素,再用溶剂洗去乙腈用于下一批次反应。
量取1.5mL乙腈溶液于玻璃瓶中,并加入525mmol/L过氧化脲或60mmoL/L过氧乙酸,最后加入30mg Novozym435,30℃、250r/min摇动若干时间后取出上清液,用乙腈-水溶液(V/V=9/1)洗净,冻干酶颗粒后用于测量酶活。定义单位时间内酶催化丙酸乙酯过水解生成1mmoL过氧丙酸所需的酶量为一个酶活单位[22]。
1.2酯类的过水解反应与α-蒎烯环氧化反应
量取1.5mL丙酸乙酯、乙酸乙酯、乙酸戊酯、己酸乙酯于玻璃瓶中并加入过氧化脲(1.1mmoL),预热至30℃后加入50mg酶,250r/min下启动反应,并于指定时间取样分析过酸浓度。
α-蒎烯环氧化反应:将25mL乙酸乙酯、丙酸乙酯、乙酸丁酯与6mmol过氧化脲充分混合,并加入60mg酶颗粒启动反应(50℃、250r/min),2h后取上清液20mL置于25mL玻璃瓶中,加入α-蒎烯(164mmol/L),于30℃、40℃、50℃、60℃反应若干时间后取出样品气相分析。
1.3以丙酸乙酯为溶剂的酶催化过水解机制
将25mL丙酸乙酯(0.5mol/L)乙腈溶液和6.25mmol过氧化脲(UHP)充分混合,取上清液20mL(碘量法测量体系中初始H2O2浓度并计算得出H2O2摩尔数)置于25mL的玻璃瓶中,加入60mg酶颗粒启动反应(30℃、250r/min),取样分析过酸浓度。为了考察水对过水解反应的影响,在反应玻璃瓶中加入一定量水,使得H2O与H2O2摩尔比为1∶10,其他反应过程和条件相同,测定过酸生成量。同时设置以丙酸(0.5mol/L)为酰基供体的对照实验。
1.4α-蒎烯环氧化反应转化率、收率以及过酸浓度的测定
α-蒎烯以及其环氧化物浓度用气相色谱(美国安捷伦公司,型号6890)测定。色谱条件:分流比为20/1,进样口与检测器分别设定为220℃和230℃。升温程序:初温100℃,以15℃/min的速率升至160℃。色谱柱为Hp-1(Agilent,30m×0.25mm×0.25μm),正十二烷[>99.0%(GC),Aladdin]为内标物。
反应转化率与收率的计算公式如式(1)、式(2)。

式中,C0为初始加入的α-蒎烯的浓度,g/L;C残为样品中残留的α-蒎烯的浓度,g/L;C1为样品中2,3-环氧蒎烷的浓度,g/L。活性氧原子的浓度用碘量法测量,剩余过氧化氢浓度用铈量法测定,过酸浓度便是两者的差值[23-24]。
2 结果与讨论
2.1不同酯类对反应的影响
表1反映了不同酰基供体对环氧化反应的影响。首先不同酯的物化特性会影响H2O2在有机相中的传质,一般来说, lgP(P是溶剂在正丁醇与水中的分配系数,表征疏水性)值较低的溶剂中H2O2浓度较高,这有利于推动环氧化反应,如乙酸戊酯体系中H2O2浓度仅有14mmol/L,其3h的转化率为39%;而乙酸乙酯时H2O2浓度可达到80mmol/L,转化率可达到86%。但是过高的H2O2浓度可能造成酶的失活[25],存在底物抑制作用。以乙酸甲酯为例,尿素过氧化氢在此系统中不稳定,短时间内释放出大量过氧化氢,虽然有机相中H2O2浓度在反应中能保持在400mmoL/L以上,但其3h的转化率只有44%。

表1 不同酰基供体对催化α-蒎烯反应的影响
其次,由于酯同时也是过水解反应底物,酯类分子结构可能影响过水解反应,进而影响总反应速率。如在己酸乙酯中,H2O2浓度为24mmol/L,3h的转化率为28%,而乙酸戊酯中H2O2浓度仅为14mmol/L,但其转化率却可达到39%。
从反应速率考虑,丙酸乙酯和乙酸乙酯作为酰基供体是较好的选择。
2.2酯类对反应稳定性的影响
进一步选择乙酸乙酯和丙酸乙酯为溶剂,考察酶的稳定性,结果如图2所示。虽然两种溶剂中第一批次反应的转化率基本相同,但是在丙酸乙酯中酶批次稳定性明显好于乙酸乙酯:前者第七批次时的转化率仍达到35%,而后者的转化率只剩下4%,分析原因认为与反应中的副产物有关,乙酸乙酯的过水解产物——过氧乙酸和环氧化反应后的产物——乙酸,相较于丙酸乙酯来说碳链都更短,在相同催化时间内对酶的毒害更大[8,26];而且乙酸乙酯体系中H2O2浓度也更高。因此可以看出丙酸乙酯作为溶剂和酰基供体较为合适。在ANKUDEY 等[11]研究中,乙酸乙酯体系中Novozym 435催化烯烃环氧化连续使用6批次而没有显著酶活损失,可能是由于高用酶量(50mg/1.5mL)。

图2 以丙酸乙酯、乙酸乙酯为酰基供体时反应稳定性
一般认为,H2O2和过酸是环氧化体系中酶失活的主要因素[8]。为此分别研究了脲素、溶剂、α-蒎烯、H2O2和过酸等因素对酶活稳定性的影响,发现脲素、溶剂、α-蒎烯对酶活稳定性影响不大;但是与文献报道不同的是,如图3所示,本研究体系中H2O2和过氧乙酸对酶活的影响也不显著,酶分别保存在525mmol/L H2O2和60mmol/L过氧乙酸的体系中,失活曲线基本一致,900min后仍能保持90%的活力。但是当将酶保存在过氧乙酸与H2O2共存的体系中,可以发现此时酶失活加剧,400min剩余酶活仅有60%,900min后仅剩余12%,这说明此环氧化体系中酶的失活可能是过酸与H2O2的协同作用,这有待于进一步研究。

图3 过酸与H2O2对酶活的影响
2.3不同酰基供体对酶催化产过酸以及α-蒎烯环氧化反应的影响
为探究不同酯类如何影响过水解反应以及Chemical epoxidation反应速率进而控制整个Chemo-enzymatic epoxidation反应,首先测定其过水解速率,如图4所示。
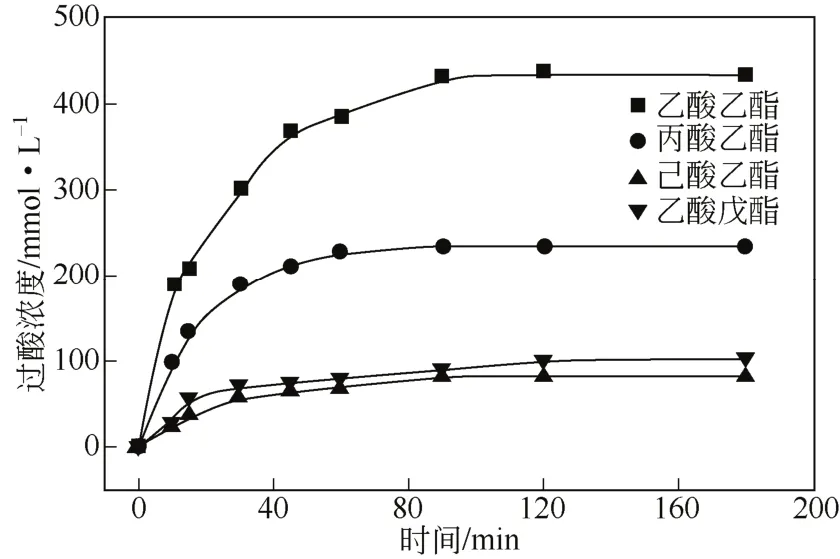
图4 酶催化不同酯过水解
以4种酯为酰基供体的过水解反应几乎均在60min后达到平衡;反应初期,酶催化乙酸乙酯过水解速率最大,丙酸乙酯次之,乙酸戊酯稍大于己酸乙酯,其过酸平衡浓度依次递减,分别为434mmol/L、234mmol/L、101mmol/L和82mmol/L。可以发现,过水解反应与表1中环氧化反应结果并不严格对应,虽然以丙酸乙酯和乙酸乙酯为底物的生成过酸平衡浓度相差两倍(其H2O2浓度基本相同),但是α-蒎烯总环氧化速率在这两种溶剂中相近;而己酸乙酯过水解速率稍小于乙酸戊酯,其环氧化速率也较小。这是由于α-蒎烯化学环氧化反应也影响着总环氧化速率。为此,测定以乙酸乙酯、丙酸乙酯、乙酸丁酯为酰基供体时的环氧化反应进程,如图5所示,环氧化速率都随温度升高而增大,如丙酸乙酯体系中30℃时120min转化率为63%,60℃转化率增至72%。在同一温度下,乙酸乙酯体系中α-蒎烯转化较快,50℃时反应30min乙酸乙酯体系中转化率达到59%,而丙酸乙酯体系与乙酸丁酯体系中转化率为46%、51%,3种体系中反应速率差别不大,可能是由于过酸初始浓度不同所致。

图5 不同温度下丙酸乙酯体系、乙酸乙酯体系、乙酸丁酯体系中蒎烯烃反应速率
因此,以速率常数来表征不同酯对反应速率的影响更为理想。根据文献报道[21],蒎烯chemical epoxidation反应符合二级动力学,拟合方程为式(3)。
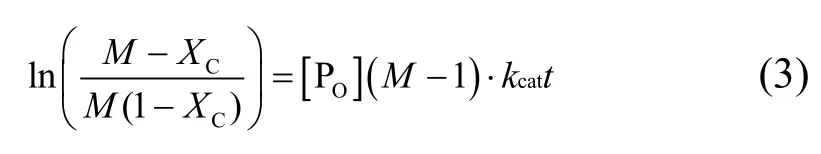
式中,M=[Q]]/[PO];[PO]为α-蒎烯初始浓度;QO为过酸初始浓度;XC为α-蒎烯转化率。

图6 不同酯体系中蒎烯环氧化反应拟合曲线
通过图6与拟合方程可得出不同温度下化学环氧化反应速率常数,如表2所示。
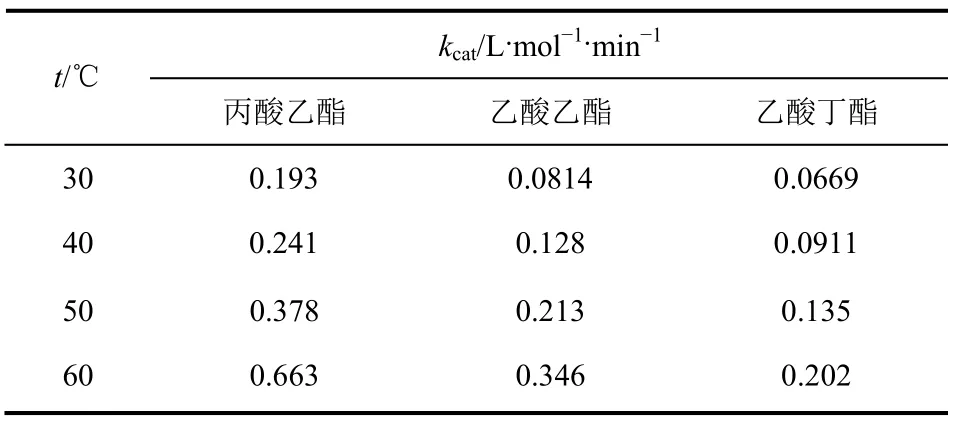
表2 不同酯体系蒎烯环氧化反应速率常数
3种体系中速率常数都随温度升高而变大。而同一温度下不同酯体系中蒎烯化学环氧化反应速率不同,首先是由于不同碳链长度过氧酸与烯烃反应时由于其质子化状态不同而速率相异[27],如表2中丙酸乙酯体系环氧化常数大于乙酸乙酯体系,这与前述丙酸乙酯体系过水解速率较小而两种酰基供体体系总环氧化速率相同的结果对应;其次,表2中乙酸乙酯体系与乙酸丁酯体系中反应物都为过氧乙酸,但乙酸乙酯体系速中率常数较大,应该是由溶剂效应引起的,即溶剂自身溶解底物的特性。
2.4以丙酸乙酯为溶剂的酶催化过水解反应机制
过水解反应一般被认为是环氧化过程的限速步骤,探究丙酸乙酯体系中酶催化机制有助于了解整个环氧化过程。
图7反映了在有机体系中以酸或酯为酰基供体以及外加水量对酶催化过水解效率的影响。可以看出,在无外加水的环境中丙酸乙酯过水解反应速率最大,在180min内过酸可达到59.2mmol/L的平衡浓度;而以丙酸为酰基供体的过水解反应无论是反应速率还是生成的过酸平衡浓度均比以丙酸乙酯为低,过酸平衡浓度只有7.5mmol/L,这说明了在有机体系中以酯为酰基供体要优于丙酸。由此可见,当选用丙酸乙酯作为酰基供体与溶剂时,作为过水解产物之一的丙酸基本不能被酶利用再生成过酸。

图7 酰基供体与H2O对产过酸的影响
当外加水量为240mmol/L时,过丙酸平衡浓度下降至48.7mmol/L,而当c(H2O)=10c(H2O2)= 2400mmol/L时,平衡浓度下降至32.2mmol/L,进一步考察上述体系中丙酸乙酯的消耗,当加入的H2O从0增至2400mmol/L时,丙酸乙酯转化率由30%增至88%,表明当体系中同时存在H2O和H2O2时,酯的水解和过水解反应同时进行,是一对竞争性反应,其次,酯的水解部分由于生成的丙酸不能再次转化为过酸而“无效率”,因而外加水会降低过酸平衡浓度。故以丙酸乙酯作为酰基供体时,有机环境可以减少水解反应的发生,因此更适合于环氧化反应。需要指出的是,各个体系中总的活性氧原子浓度(240mmol/L左右,即过酸与剩余过氧化氢浓度之和)在反应过程中都基本不变,这说明生成的过酸和过氧化氢在体系中能保持稳定。
这与XU等[10]以己内酯为溶剂和酰基供体的环氧化反应机制不同:当H2O和H2O2同时存在于有机相时,作为产物之一的6-羟基己酸仍可作为酰基供体在酶催化作用下生成过酸,而且其过水解速率是酯的60倍。
3 结 论
对于α-蒎烯的环氧化过程,以酯作为溶剂与过水解底物、采用过氧化脲为氧源的有机体系可以获得较高的收率。以反应速率和酶批次稳定性两个指标来优化酯的种类、温度等因素,发现丙酸乙酯或乙酸乙酯中可以获得最佳的环氧化反应转化率,批次使用稳定性则以丙酸乙酯为佳;反应转化率和收率在30℃和40℃之间没有明显差别,但30℃时酶可以获得更好的稳定性。在最优条件下采用30mg/L的酶量下3h的环氧转化率为87%,6批次反应后仍能达到47.6%。对反应机制的研究显示,酯的不同会显著影响有机相中H2O2的浓度、过酸平衡浓度等,进而影响总环氧化反应速率;在丙酸乙酯体系中酶只能以酯为酰基供体产过酸,丙酸基本不能再被利用,而且丙酸乙酯的水解反应和过水解反应为竞争性反应,因此有机体系更有利于反应。
参考文献
[1] BJORKLING F,GODTFREDSEN S E,KIRK O. Lipase-mediated formation of peroxycarboxylic acids used in catalytic epoxidation of alkenes[J]. Journal of the Chemical Society,Chemical Communications,1990(19):1301-1303.
[2] BERNHARDT P,HULT K,KAZLAUSKAS R J. Molecular basis of perhydrolase activity in serine hydrolases[J]. Angewandte Chemie,2005,44(18):2742-2746.
[3] KAPOOR M,GUPTA M N. Lipase promiscuity and its biochemical applications[J]. Process Biochemistry,2012,47(4):555-569.
[4] RANGARAJAN B,HAVEY A,GRULKE E A,et al. Kinetic parameters of a two-phase model forin situ epoxidation of soybean oil[J]. J. Am. Oil. Chem. Soc.,1995,72(10):1161-1169.
[5] PIERRE V,AOUF C V C F,FULCRAND H,et al. The use of lipases as biocatalysts for the epoxidation of fatty acids and phenolic compounds[J]. Green Chemistry,2014,16(4):1740-1754
[6] CHUA S C,XU X,GUO Z. Emerging sustainable technology for epoxidation directed toward plant oil-based plasticizers[J]. Process Biochemistry,2012,47(10):1439-1451.
[7] ORELLANA-COCA C,CAMOCHO S,ADLERCREUTZ D,et al. Chemo-enzymatic epoxidation of linoleic acid:parameters influencing the reaction[J]. European Journal of Lipid Science and Technology,2005,107(12):864-870.
[8] TORNVALL U,ORELLANA-COCA C,HATTI-KAUL R,et al. Stability of immobilized Candida antarctica lipase B during chemo-enzymatic epoxidation of fatty acids[J]. Enzyme and Microbial Technology,2007,40(3):447-451.
[9] 刘元法,王兴国,员克志. 脂肪酶催化制备环氧棉籽油的研究[J].中国油脂,2007(1):39-42.
[10] XU Y,KHAW N R B J,LI Z. Efficient epoxidation of alkenes with hydrogen peroxide,lactone,and lipase[J]. Green Chemistry,2009,11(12):2047.
[11] ANKUDEY E G,OLIVO H F,PEEPLES T L. Lipase-mediated epoxidation utilizing urea-hydrogen peroxide in ethyl acetate[J]. Green Chemistry,2006,8(10):923-926.
[12] MONTEIRO J,VELOSO C. Catalytic conversion of terpenes into fine chemicals[J]. Topics in Catalysis,2004,27(1/4):169-180.
[13] BICAS J L,DIONÍSIO A P,PASTORE G U M. Bio-oxidation of terpenes:an approach for the flavor industry[J]. Chemical Reviews,2009,109(9):4518-4531.
[14] SKOURIDOU V,STAMATIS H,KOLISIS F N. Lipase-mediated epoxidation of α-pinene[J]. Journal of Molecular Catalysis B:Enzymatic,2003,21(1/2):67-69.
[15] SKOURIDOU V,STAMATIS H,KOLISIS F N. A study on the process of lipase-catalyzed synthesis of α-pinene oxide in organic solvents[J]. Biocatal Biotransform,2003,21(6):285-290.
[16] TZIALLA A A,PAVLIDIS I V,FELICISSIMO M P,et al. Lipase immobilization on smectite nanoclays:characterization and application to the epoxidation of α-pinene[J]. Bioresource Technology,2010,101(6):1587-1594.
[17] TZIALLA A A,KALOGERIS E,ENOTIADIS A,et al. Effective immobilization of Candida antarctica lipase B in organic-modified clays:application for the epoxidation of terpenes[J]. Materials Science and Engineering:B,2009,165(3):173-177.
[18] 尹延柏,宋湛谦,商士斌,等. 环氧蒎烷异构生成龙脑烯醛研究进展[J]. 现代化工,2007(7):23-27.
[19] RÜSCH GEN KLAAS M,WARWEL S. Chemoenzymatic epoxidation of alkenes by dimethyl carbonate and hydrogen peroxide[J]. Organic Letters,1999,1(7):1025-1026.
[20] WARWEL S. Chemo-enzymatic epoxidation of unsaturated carboxylic acids[J]. Journal of Molecular Catalysis B:Enzymatic,1995,1(1):29-35.
[21] YADAV G D,BORKAR I V. Kinetic modeling of microwave-assisted chemoenzymatic epoxidation of styrene[J]. AIChE Journal,2006,52(3):1235-1247.
[22] YADAV G D,DEVI K M. Enzymatic synthesis of perlauric acid using Novozym 435[J]. Biochemical Engineering Journal,2002,10 (2):93-101.
[23] GREENSPAN F P,MACKELLAR D G. Analysis of aliphatic per acids[J]. Analytical Chemistry,1948,20(11):1061-1063.
[24] SWERN D. Differential analysis of hydrogen peroxide,peroxy acid and diacyl peroxide on a single sample[J]. Org Peroxides,1970,501:1.
[25] HAGSTROM A E,TORNVALL U,NORDBLAD M,et al. Chemo-enzymatic epoxidation-process options for improving biocatalytic productivity[J]. Biotechnology Progress,2011,27(1):67-76.
[26] ORELLANA-COCA C,BILLAKANTI J M,MATTIASSON B,et al. Lipase mediated simultaneous esterification and epoxidation of oleic acid for the production of alkylepoxystearates[J]. Journal of Molecular Catalysis B:Enzymatic,2007,44(3/4):133-137.
[27] SHI H,ZHANG Z,WANG Y. Mechanism on epoxidation of alkenes by peracids:a protonation-promoted pathway and its quantum chemical elucidation[J]. Journal of Molecular Catalysis A:Chemical,2005,238(1/2):13-25.
综述与专论
Chemo-enzymatic epoxidation of α-pinene:ester as influencing factors and mechanism of the reaction
XIONG Yang1,QIN Yimin2,TANG Aixing1,WEI Huilin1,LIU Youyan1,2
(1College of Chemistry and Chemical Engineering,Guangxi University,Nanning 530004,Guangxi,China;2Guangxi Key Laboratory of Biorefinery,Nanning 530003,Guangxi,China)
Abstract:Chemo-enzymatic epoxidation of α-pinene was achieved in a non-aqueous system which employed esters as solvent and perhydrolysis substrate,urea-hydrogen peroxide (UHP) as an oxygen source and lipase as catalyst. Different esters were found to affect the reaction significantly. The optimal reaction condition was 30℃ and ethyl propionate as the solvent considering both the reaction rate and the stability of enzyme. Under these conditions,the conversion could achieve 87% after 3h and remain 47.6% after 6 cycles. The synergy toxic effect of H2O2and peracid was more significant than that of single one. It was found that using different acyl donors such as ethyl acetate,ethyl propionate,amyl acetate,ethyl caproate and the like,the reaction of lipase-catalyzed production of peracid can all reach equilibrium within 60min. However,the equilibrium concentration of peracid and the rate of chemical epoxidation greatly depended on the ester employed,which in turn affected the overall epoxidation of α-pinene. The mechanism of the reaction with ethyl propionate as the exclusive acyl donor was discussed. The reaction rate and the equilibrium concentration for producing peracid by enzyme with propionic acid was significantly lower than those with esters and it was found that there are competitions between the enzymatic hydrolysis with H2O and the perhydrolysis with H2O2whenbook=221,ebook=228H2O existed,and therefore the nonaqueous environment was more suitable for the epoxidation.
Key words:ethyl propionate; α-pinene; epoxidation; deactivation; lipase; perhydrolysis; biocatalysis
基金项目:国家自然科学基金(21276053)及广西自然科学基金(0991001)项目。
收稿日期:2015-04-28;修改稿日期:2015-06-30。
DOI:10.16085/j.issn.1000-6613.2016.01.030
中图分类号:Q 503
文献标志码:A
文章编号:1000–6613(2016)01–0220–07
