异丙肾上腺素在硅酸铋离子交换薄层上的选择性分离与测定
2015-12-26VanikGHOULIPOUR,ZahraHASSANKHANI-MAJD
The increasing use of drugs by athletes to enhance athletic performance has become a matter of serious concern for sport authorities[1]. The doping analysis is viewed as an effective deterrent. The similarity in chemical behaviors and the structures of doping drugs demands selective separation procedures. The chromatographic techniques are effectively used in anti-doping control laboratories[2-5]. Due to the high selectivity of new synthetic inorganic ion-exchangers,their use in thin layer chromatography (TLC)has transformed it into a fast and powerful technique for separation of identical compounds[6,7].
Isoproterenol,a prohibited stimulant,is used by some athletes to enhance their performance[8],although it has very harmful effects on human body[9-11]. Isoproterenol hydrochloride is 3,4-dihydroxy-α-[(isopropylamino)methyl]benzyl alcohol hydrochloride,a synthetic sympathomimetic amine that is structurally related to epinephrine but acts almost exclusively on beta receptors. The representative molecular structure of this amine is given in Fig.1. Isoproterenol is a catecholamine drug which has harmful effects on heart,bronchial and gastrointestinal muscles.Therefore its selective determination is very important.

Fig.1 Chemical structure of isoproterenol
Various methods including gas chromatography[12],chemiluminescence[13-16],spectrophotometry[17-20],electrochemical methods[21,22]and HPLC[23]have been used for detection of isoproterenol. In continuation to our studies on doping drugs [24],we have developed a rapid and selective high performance thin layer chromatography (HPTLC)method for separation and determination of isopreterenol in presence of other doping drugs on thin layers of bismuth silicate inorganic ion-exchanger. The results obtained by this method agreed with those obtained by the official method[25]and therefore can be used for selective determination of isoproterenol in the drug dosage,blood plasma and urine samples.
1 Experimental
1.1 Apparatus and chemicals
A Camag automatic TLC coater was used to prepare ion-exchange plates. The development was performed in a twin-trough chamber (Camag)for 20 cm×20 cm plates. The sample and standard solutions were applied on plates by means of a Camag auto sampler III. The sample and standard zone areas were measured by using a Camag TLC Scanner-3. All the chemicals and reagents were of analytical grade (Merck and BDH). Isoproterenol (98%)was purchased from Sigma-Aldrich (Germany). The drugs studied were amphetamine (99%), caffeine (99%),ephedrine (98%),methylendi-oxyamphetamine(98%),methadone (98%),pentazocine (98%),pethidine (98%),bemegride (99%),pemoline(99%),strychnine (98%)and salbutamol (96%)(Sigma-Aldrich),chlorphentermine and ethylamphetamine (certified reference materials grade)(United Nations Office for Drug Control and Crime Prevention).
1.2 Standard solutions
A stock standard solution (1 000 μg/mL)of isoproterenol was prepared by dissolving isoproterenol (0.100 0 g)in methanol (100 mL). Quantitative standards for calibration plot were prepared by appropriate dilution of the stock solution. The solutions of other drugs (15 μg/mL)were also prepared in methanol.
1.3 Chromatography
1.3.1 Preparation of ion-exchange plates
A solution of bismuth nitrate (0.1 mol/L,500 mL)in 2 mol/L HNO3and a solution of sodium silicate (0.1 mol/L in Si,500 mL)in 2 mol/L NaOH were mixed dropwise with constant stirring in a flat-bottomed flask till the content of the flask was just neutral to methyl red indicator. The reaction mixture was left to coagulate overnight and the white gel formed was washed five times with distilled water by decantation until the supernatant was free from ions. The supernatant was completely removed. A slurry prepared by mixing the gel (75 mL)with silica gel G 60 powder (14 g)as binder. The slurry was then poured immediately into the automatic TLC coater to coat seven 20 cm×20 cm glass plates with a 300 μm layer thickness[26]. The plates were dried in an oven at 60-70 ℃for 2 h and then stored at room temperature inside a incubator.
1.3.2 Procedure
Standard and sample solutions (1 000 nL)were applied automatically (in spray mode)by means of Camag auto sampler as 2 mm bands,with band velocity of 10 mm/s. The distance between bands was 14.7 mm;the distance from the plate edge was 170 mm,and the distance from the bottom of the plate was 30 mm on bismuth silicate ion-exchanger plates. The plates were developed upto 12.5 cm from the origin with a mixture of methanol and 0.1 mol/L formic acid (3 ∶7,v/v)as the mobile phase inside a TLC chamber. The development time was approximately 32 min. After the development,the mobile phase was evaporated from the plate by drying in a fumehood for 10 min. Under these conditions the RTand RL(RTis the retention factor of rear of spot;RLis the retention factor of leading of spot)values of isoproterenol were 0.40-0.46,while the retention factor (Rf)of the other drugs were lower or higher than this value.
1.3.3 Detection reagents
Isoproterenol was located with a fresh solution of 10 g/L silver acetate prepared in distilled water. The detection reagent for other drugs was iodine solution which was prepared by dissolving 2 g iodine and 3 g potassium iodide in demineralized water (100 mL).
1.3.4 TLC analysis
The sample and standard zone areas were measured by using Camag TLC Scanner-3 in reflectance-absorbance mode with a tungsten lamp set at 410 nm,with slit length of 4 mm,slit width of 0.45 μm,scanning rate of 5 mm/s and the monochromator bandwidth of 5 nm. The CATS-3 software controlling the densitometer produced a linear regression calibration curve relating the standard zone weights to their scan areas and interpolating the sample zone weights.
1.4 Applicability of the method
1.4.1 Analysis of dosage forms
The contents of five commercial injection vials containing isoproterenol were mixed well,transferred into a 50 mL measuring flask and filled up to the mark. The procedure described above for the pure sample was followed. The nominal content was calculated from the corresponding regression equation.
1.4.2 Determination of isoproterenol in spiked human plasma
The plasma samples obtained from healthy persons were supplied by Iranian Blood Transfusion Organization (Tehran,Iran). To 2 mL plasma sample contained in a centrifuge tube was added 2 mL of isoproterenol standard solution (250 μg/mL),then mixed well using a vortex mixer and the proteins were precipitated out by adding 4 mL acetonitrile. The mixture was centrifuged at 2 400 r/min for 5 min. The supernatant was completely transferred into 10 mL measuring flask and was filled with distilled water till mark,then the above described procedure was followed.
1.4.3 Determination of isoproterenol in spiked urine samples
Fresh urinary samples were taken from healthy volunteers in the laboratory. The volunteers had abstained from any medications during the week preceding the study. To 6 mL of urine sample was added 2 mL of isoproterenol standard solution(250 μg/mL),then 2 mL methanol was added for the subsequent removal of proteins. These samples were centrifuged for 3 min at 6 000 r/min.Then the upper liquid was removed and the procedure described for standard sample was followed.
2 Results and discussion
The results show that fast separation of isoproterenol from other doping drugs has been achieved on thin layer of bismuth silicate using a mixture of methanol and 0.1 mol/L formic acid aqueous solution (3 ∶7,v/v)as the mobile phase. The effectiveness of the anion-exchanger bismuth silicate for the development of rapid and selective separations of drugs are based on selective hydrolytic adsorption and ion exchange process on the thin layer bed not only through the electrostatic exchange but also through the varying sorption of negative,neutral and positive charge species of drugs formed in mobile phase.
The direct estimation of separated isoproterenol zone on the ion-exchanger plate by the help of Camag TLC Scanner provides a fast and selective method for its determination. Calibration curves were drawn by the external standard technique following linear regression analysis,plotting concentrations against peak areas[27]. The calibration curve for each plate obtained from the results of six standard bands was linear for isoproterenol ranging from 10 to 60 ng per spot,with the correlation coefficients (R2)ranging from 0.987 1 to 0.998 8. The adequacy of the regression model was examined by plotting the residuals[28]. The residual data were distributed randomly around the zero line,which confirmed the correctness of calibration plot[29]. The accuracy of this method was also determined with certified reference samples. Two groups of samples,each including 12 test solutions containing 15 and 25 μg/mL isoproterenol and 15 μg/mL other drugs were prepared and the analysis was done in triplicate by means of a system calibration chromatography[29,30]. The whole analytical procedure was repeated three times using the same chromatographic plates[31]. The recoveries were 99.10%for 15 ng per spot and 100.34% for 25 ng per spot,showing the high accuracy of this procedure with good repeatability.
In accordance with ICH (International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use)guideline Q2B repeatability was determined by repeated analysis of a homogeneous sample using the same analytical procedure and the same equipment in the same laboratory and calculating the standard deviations[27]. Intermediate precision was determined by analyzing samples on different plates [32]. For further evaluation of the precision,standard deviation of the method based upon the pooled precision data was calculated for two groups of samples and the relative standard deviation (RSD)of the recoveries. The average amount of isoproterenol found for one group of samples was 14.86 ng and the RSD was 2.93%;for another group of samples the average amount was 25.09 ng and the RSD was 2.83%. The limit of detection (LOD)and the limit of quantification(LOQ)were determined by the standard deviation (SD)method. The blank samples were applied in triplicate,and the peak area of this blank was calculated. The LOD and LOQ were determined from the slope of the calibration plot and the SD of the responses for the blank sample using the formulas LOD =2×SD/slope and LOQ =10×SD/slope [31,32]. LOD was equal to 7.7 ×10-7mol/L. LOQ was 3.85×10-6mol/L.
For the six consecutive periods,this developed HPTLC method was applied for the determination of isoproterenol in commercial formulations. Isoproterenol is commercially available in the form of injection ampoule. The results indicate that the method developed can also be used for the quality control of isoproterenol (Table 1).

Table 1 Assay results for the HPTLC determination of isoproterenol (isuprel)in commercial ampoules (n=6)
This developed method was successfully applied for the determination of isoproterenol in human blood plasma and urine samples (Table 2).The recovery,accuracy and reproducibility of the method were checked statistically. As shown in Fig.2 and 3,the method has sufficient specificity and selectivity as isoproterenol was well separated and the Rfof excipients proved no interference (peak overlapping)with isoproterenol. The blank plasma and urine did not interfere with the assay.

Table 2 Quantitative separation of isoproterenol from blood plasma and urine samples (n=6)
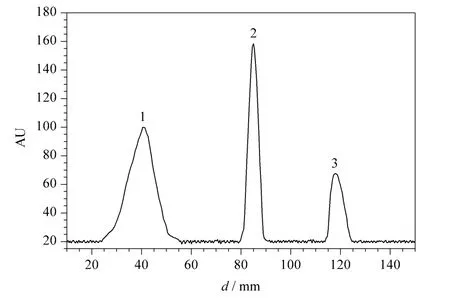
Fig.2 Chromatogram obtained from a blood plasma sample spiked with isoproterenol

Fig.3 Chromatogram obtained from a urine sample spiked with isoproterenol
The purpose of this study was to develop a time saving,cost effective,simple,precise and sensitive method for the determination of isoproterenol which does not require extensive preliminary sample treatment. The developed HPTLC method meets these requirements. The comparision of the present work with other published methods is summarized in Table 3. The LOD for this work is lower than those of the HPLC and chemiluminescence methods which require solid phase microextraction and derivatization for sample preparation in comparision with our work which is very simple and inexpensive. This developed method has suitable accuracy and precision for routine estimation of isoproterenol in various samples. In addition,the presented HPTLC method is simpler than those described in the literature with minimum toxic effluent generation in accord to greener analytical chemistry[36].
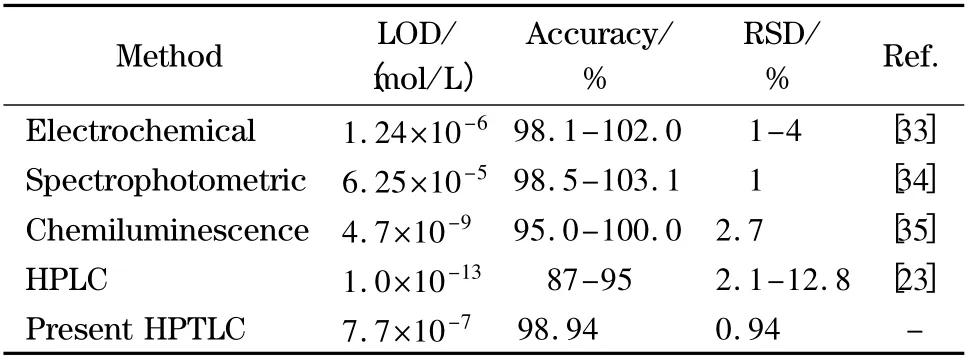
Table 3 Comparision of LODs,accuracies and RSDs of various methods for isoproterenol determination
3 Conclusion
A fast and selective HPTLC method for the separation and determination of isoproterenol employing thin layers of bismuth silicate ion-exchanger has been developed. Good sensitivity,low LOD and high selectivity of this method show its potential applications for determination of isoproterenol in real samples such as ampoule,urine and plasma. The advantage of TLC is the high sample throughput which results from the small amounts of sample preparation required and the simultaneous quantification of several samples with a short analysis time and low solvent consumption. Moreover,with a single plate up to 12 chromatographic runs could be performed simultaneously.
The authors are grateful to the Dean of the Faculty of Chemistry for providing research facilities.
[1] Conway L,Morgan D. Drugs in Sports. London:British Medical Association,2002
[2] Jimnez C,Ventura R,Segura J. J Chromatogr B,2002,767:341
[3] Mueller R K,Grosse J,Lang R,et al. J Chromatogr B,1995,674:1
[4] Sherma J,Fried B. Handbook of Thin-Layer Chromatography. New York:Marcel Dekker Inc.,1991:407
[5] Sherma J. Anal Chem,2010,82:4895
[6] Qureshi M,Varshney K G. Inorganic Ion-Exchangers in Chemical Analysis. Florida:CRC Press,1991
[7] Husain S W,Ghoulipour V,Sepahrian H. Acta Chromatogr,2004,14:102
[8] Bowers L D,Podraza J. USADA Guide to Prohibited Classes of Substances and Prohibited Methods of Doping. Colorado:Anti-Doping Agency,2002:7
[9] Shaw P A,Yu W A. Life Sci,2001,70:301
[10] Nieto J L,Laviada I D,Guillen A,et al. Cell Signal,1993,5:169
[11] Krenek P,Kmecova J,Kucerova D,et al. Eur J Heart Fail,2009,11(12):140
[12] Watson J R,Lawrence R C. J Pharm Sci,1977,66:560
[13] Liu Y M,Cao J T,Zheng Y L,et al. J Sep Sci,2008,31(13):2463
[14] Zhou G J,Zhang G F,Chen H Y. Anal Chem Acta,2002,463:257
[15] Zhang C,Huang J,Zhang Z,et al. Anal Chem Acta,1998,374:105
[16] Al-Warthan A A,Al-Tamrah S A,Al-Akel A. Anal Sci,1994,10:449
[17] Bonifacio Y G,Marccoline-Junior L H,Fatibello-Filho O.Anal Lett,2004,37(10):2111
[18] Lupetti K O,Vieira I C,Fatibello-Filho O. Talanta,2002,57:135
[19] Solieh P,Polydorou C K,Koupparis M A,et al. J Pharm Biomed Anal,2000,22:781
[20] Nevado J J B,Gallego J M L,Laguna P B. Anal Chem Acta,1995,300:293
[21] Ensafi A A,Khoddami E,Karimi-Maleh H. Int J Electrochem Sci,2011,6:2596
[22] Kutluay A,Aslanoglu M. Acta Chim Slov,2010,57:157
[23] Elord J L,Schmit J L,Morley J A. J Chromatogr A,1996,723:235
[24] Hassankhani-Majd Z,Ghoulipour V,Husain S W. Acta Chromatogr,2006,16:173
[25] Ahuja S,Scypinski S. Handbook of Modern Pharmaceutical Analysis. Burlington:Academic Press,2011:430
[26] Ghoulipour V,Husain S W. Anal Sci,2000,16:1079
[27] ICH Guidelines Q2B,Validation of Analytical Procedures:Methodology (CPMP/ICH/281/95). Geneva,1996
[28] Papp E,Farkas A,Otta K H,et al. J Planar Chromatogr,2000,13:328
[29] Ferenczi-Fodor K,Vegh Z,Nagy-Turak A,et al. J AOAC Int,2001,84:1265
[30] Dallas F A A,Read H,Ruane R J,et al. Recent Advances in Thin-Layer Chromatography. New York: Springer,1988:11
[31] Ferenczi-Fodor K,Renger B,Vegh Z. J Planar Chromatogr,2010,23:173
[32] Ramirez A,Gutierrez R,Diaz G,et al. J Chromatogr B,2003,784:315
[33] Mazlom-Ardakani M,Sabaghian F,Khoshroo A,et al.Chin J Catal,2014,35:565
[34] Lupetti K O,Vieira I C,Fatibello-Filho O. Talanta,2002,57:135
[35] Rezaei B,Ensafi A A,Haghighatnia F. Anal Methods,2012,4:1753
[36] Rocha F R P,Nobrega J A,Fatibello D. Green Chem,2001,3:216
