用于XRF测量微量Np的磁助分离制样方法
2015-12-25郑维明吴继宗康海英邓惟勤邵少雄
张 彤,全 葳,郑维明,吴继宗,康海英,邓惟勤,邵少雄
1.中国原子能科学研究院 放射化学研究所,北京 102413;2.环境保护部 核与辐射安全中心,北京 100082
镎在水溶液中价态复杂多变、多价态共存,在Purex流程中走向分散。流程多处工艺点都有Np存在,需要对部分工艺点的Np含量进行测量,准确掌握Np在流程中的走向。所有工艺点中,3EU工艺点的镎含量极低,约为1.5×10-4g/L,铀质量浓度约为70g/L,ρ(U)/ρ(Np)高达4.7×105。因此,3EU中Np的测量在Purex流程中测量难度最大。
目前可用于微量镎的测量方法主要有α能谱法、液体闪烁计数法[1]、电感耦合等离子体质谱(ICP-MS)法及X射线荧光技术等。使用放射性测量方法,如α能谱法及液体闪烁计数法测量微量镎时,会受到Pu和234U的干扰,难以得到准确数值。ICP-MS[2]是一种多元素分析技术,具有极好的灵敏度和高效的样品分析能力,测量放射性物质时,需要对仪器进行封闭,但封闭难度较大,封闭后仪器性能下降;同时,使用ICP-MS的维护及维修费用较昂贵;并且样品ρ(U)/ρ(Np)过高,需要预处理降低其比例。使用X射线荧光技术,从原理上解决了Pu和234U对Np放射性测量的影响。与ICP-MS相比,X射线荧光技术原理相对简单,结构并不复杂,仪器小巧,易于与手套箱形成封闭系统;且仪器易于维护,仪器成本、运行成本及维护成本显著降低,易于在工厂推广。X射线荧光分析中主要存在两个问题:一是大量铀的干扰,二是仪器的检测下限未能达到某些工艺点测量要求。大量铀的干扰可使用TEVA萃取色层预分离解决[3]。测量液体样品时,能够降低仪器浓度检测下限的制样方法主要有蒸发法和共沉淀法。蒸发法需要对溶液加热蒸发溶剂,处理放射性溶液时易沾污。共沉淀制样方法是通过加入共沉淀剂(如Ca(OH)2、氟化镧[4]等)和絮凝剂,将Np元素全部共沉淀析出。沉淀需过滤或离心才能进一步制样完成测量,需要引入其他分离设备,操作繁琐。本工作针对经TEVA萃取色层分离后Np含量极低的溶液,拟建立磁助捕集分离制样法,使用超顺磁性固相捕集剂将溶液中Np捕集至固相中,在外加磁场作用下快速固液分离,达到捕集、分离制样的目的。制得样品直接使用X射线荧光仪器测量,经谱图数据处理后得出溶液中Np含量。通过对Np的捕集分离,降低浓度检测下限,达到测量要求。
1 实验部分
1.1 试剂和仪器
所用试剂均为市售分析纯。
氨基磺酸亚铁溶液:在50mL烧杯中称取3.0g氨基磺酸,加入20mL 0.1%硝酸(质量分数)及0.7g还原铁粉,放置过夜。不溶物过滤后,滤液用容量瓶定容至25mL,有效期为3d。
Np溶液,中国原子能科学研究院放射化学研究所提供,c(HNO3)=2mol/L,经液闪标定后Np质量浓度为22.163mg/L,使用时加入适量氨基磺酸亚铁溶液将Np价态全部调至四价。
固相捕集剂:本工作使用的固相捕集剂是通过有机物聚合,将具有超顺磁性的纳米磁颗粒和捕集剂偶联制成超顺磁性三异辛胺(TiOA)固相捕集剂,由核工业北京化工冶金研究院合成。
TM-1Fn型漩涡混合器,日本AS ONE公司;Tri-Card 2910TR型液体闪烁仪,美国Perkin Elmer公司;CIT-3000SM型能量色散X荧光分析仪,自制,质量检出下限为0.07μg,质量浓度检出下限为1.4mg/L。
1.2 实验方法
1.2.1 磁助捕集分离制样法操作流程 磁助捕集分离制样法示意图示于图1。操作流程如下:(1)在中通管底面覆盖一张mylar膜;(2)用中通管套将mylar膜固定在中通管的底面制成样品管;(3)从上部管口加入一定量固相捕集剂;(4)从上部管口加入一定量待测溶液并用浓HNO3调酸,用氨基磺酸亚铁溶液调价;(5)使用漩涡混合仪使固相捕集剂与液相混合均匀并保持一段时间;(6)在底部管口施加磁场进行快速固液分相;(7)保持磁场强度,将液相从上部管口倒出;(8)将样品管直接放置在X荧光仪上进行测量。测得信号谱图示于图2。不同元素具有不同的特征X荧光,在谱图上对应能量道数不同。数据处理时,计算如下式:
S(Np/Ag)=I(Np)/I(Ag)
式中:S(Np/Ag)是Np、Ag峰位计数率比值,表示以Ag作为内标时Np的观测值;I(Np)、I(Ag)分别为Np、Ag特征道数范围内的计数率之和。
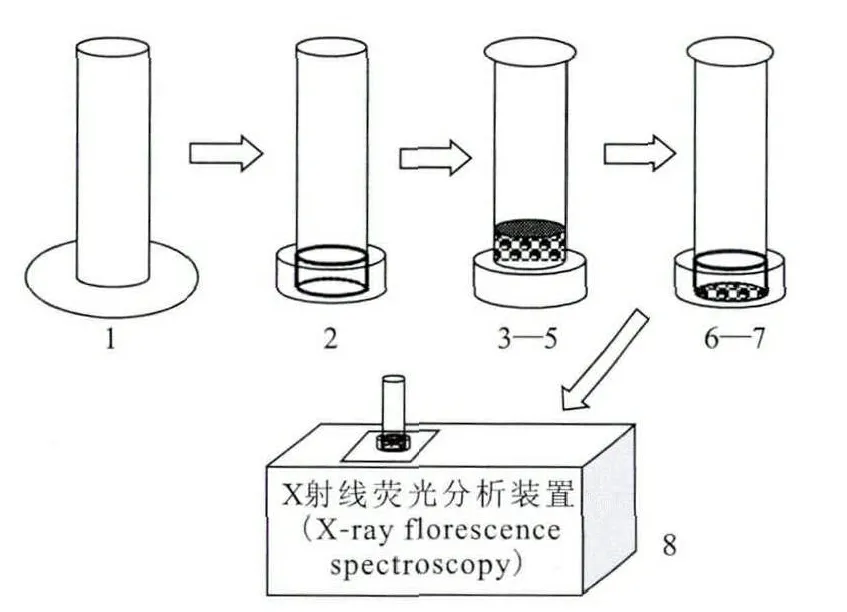
图1 磁助分离制样法示意图Fig.1 Schematic diagram for magnetically assisted sample preparation

图2 XRF测得信号谱图Fig.2 Signal spectrogram for XRF
S(Np/Ag/Fe)=I(Np)/I(Ag)/I(Fe)
式中:S(Np/Ag/Fe)为以Ag-Fe作为内标时Np的观测值;I(Fe),Fe在特征道数范围内的计数率之和。建立Np的观测值与样品溶液中Np的含量或Np浓度的工作曲线,利用该工作曲线进行测量计算。
1.2.2 磁助捕集分离制样标准样的制备 已知Np标准溶液浓度,使用液体闪烁仪测量磁助捕集分离制样后倒出上清液中Np的浓度,计算出固相颗粒中Np的质量。通过计算得到制备的Np质量分别为6.99、3.19、0.60、0.44、0.29、0.12μg的标准样及空白样品。建立Np磁助捕集分离标准样的工作曲线。依据本底3倍标准偏差原则,测得空白样品Np峰位观测值并计算其统计误差,计算检出下限。使用工作曲线计算制得样品中Np含量。固相中Np的质量与溶液中初始Np含量的比值为该制样方法的回收率。
2 结果与讨论
2.1 固相捕集剂选择
选用的固相捕集剂应具有超顺磁性、对Np具有较好的选择性。通过测量颗粒磁滞曲线筛选具有超顺磁性的四氧化三铁纳米颗粒[5]。没有外磁场时,颗粒不会表现出磁性,颗粒不团聚。底部施加外磁场时,颗粒被磁化,可迅速沉积在底面,易于固液分离制样。
通过对胺类捕集剂的筛选,选用叔胺TiOA作为捕集剂。TiOA在所有胺类捕集剂中对四价Np的选择性好,化学性能稳定,易于制备成固相捕集剂。
因此,本工作使用的固相捕集剂是将纳米级四氧化三铁颗粒和TiOA通过苯乙烯的聚合制成球形颗粒,即超顺磁性TiOA固相捕集剂。粒径范围为50~400μm,为保证捕集性能稳定,通过筛分选用粒径小于150μm的固相捕集剂用于磁助捕集分离制样。
2.2 内标物选择
磁助捕集分离制样在操作时会引入误差,对Np测量信号值有较大干扰,需要使用内标校正。制样时主要由两方面引入误差:一是固液分离时,固相颗粒中残留溶液体积不同时会影响入射光接触样品时的散射情况,改变测量信号值;二是固相颗粒在底面分布不均时,会改变入射光与样品接触情况,改变测量信号。通过对样品反加上清液体积,研究固相残留溶液体积对测量结果的影响,结果列入表1。由表1可知:在制样过程中,固相残留液相体积小于150μL,使用内标校正后,相对偏差绝对值由36%降至16%以内。
颗粒在底面分布形式主要有两种:(1)颗粒基本均匀平铺于底面;(2)颗粒聚集,偏向一侧。颗粒底面分布对测量结果的影响列入表2。由表2可知,当颗粒在管内底面分布不同时,与颗粒均匀铺满底面情况下测得结果相比,使用内标校正,相对偏差绝对值由175%降至10%以内。通过内标校正,可有效降低制样时引入的误差,提高制样稳定性。制样熟练时,使用Ag或Ag-Fe内标对结果都有较好的校正效果。
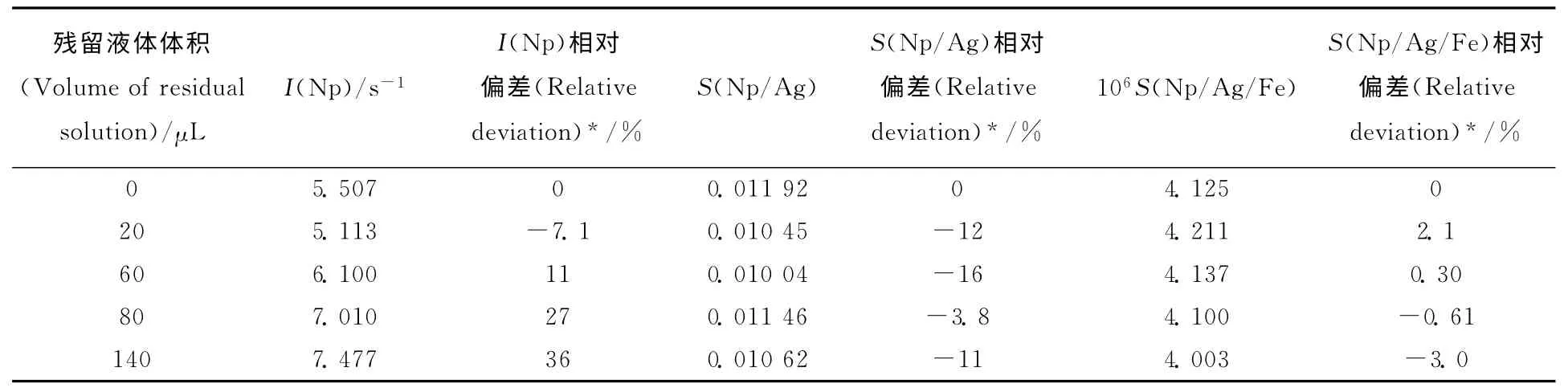
表1 固相残留溶液体积对测量结果的影响Table 1 Effects of residual solution volume in capture on Np determination
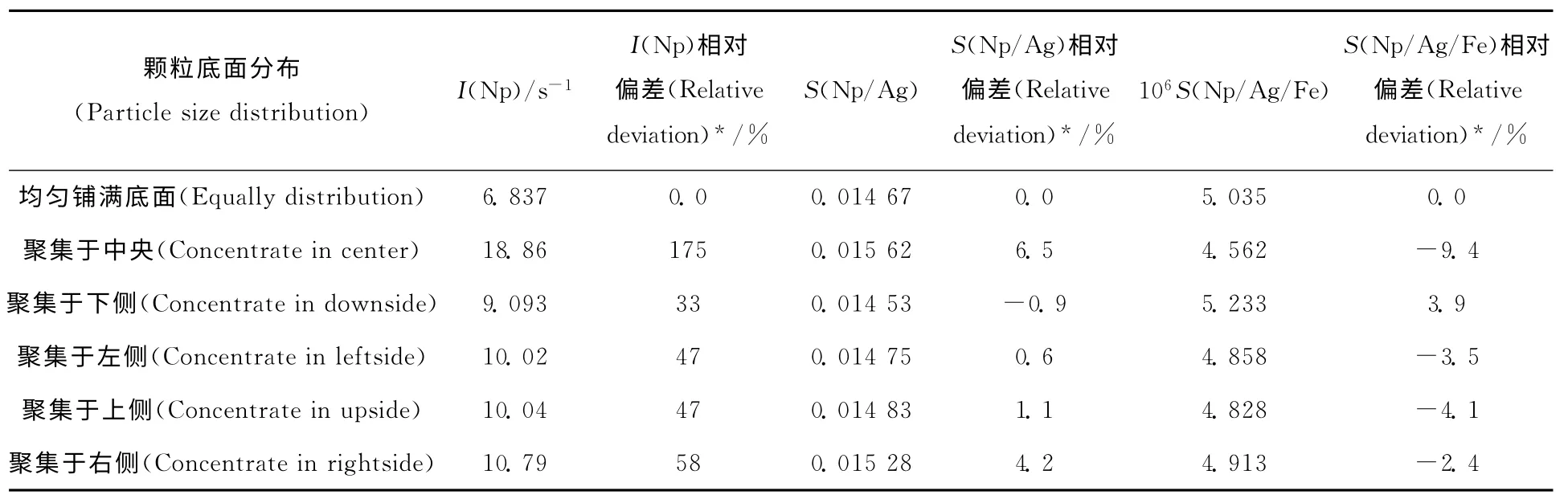
表2 颗粒底面分布对测量结果的影响Table 2 Effects of particle size distribution of capture on Np determination
2.3 反应时间对捕集的影响
在2mol/L酸度下,溶液中m(Np)=2.2μg,对Np溶液进行磁助捕集分离制样,观测值随时间的变化示于图3。由图3可知,反应8min后基本平衡,8~30min 6次测量sr=8.5%。因此,制样时取反应时间10min以保证反应平衡。
2.4 固相捕集剂用量及液相体积对捕集的影响
改变固相捕集剂用量及溶液体积,即改变相比,从而影响分配比。相比的改变同时影响颗粒在溶液中的分散状态,对固液分离制样有一定影响。在2mol/L酸度下,溶液中Np质量为2.2μg,测定不同固相捕集剂用量(m)及不同溶液体积(V)对测量结果的影响,结果列入表3。由表3可知:增加固相捕集剂用量及减少液相体积提高了Np的检测信号,但使用内标校正后,相对偏差绝对值小于15%。

图3 反应时间对测量结果的影响Fig.3 Dependence of reacting time on analysis

表3 固相捕集剂用量与溶液体积对测量的影响Table 3 Effects of the mass of capture and the volume of aqueous phase on Np determination
分析原因:增加固相捕集剂用量及减少液相体积有利于Np由液相转移至固相。但增加固相捕集剂用量时,使得测得Ag信号值增加,增加了制得样品对于入射光的散射,数据处理时需要使用Ag信号值作为内标,因此减弱了改变固液相比时对最终测量结果的影响。因此,制样时选用固相捕集剂0.1g,液相体积5mL。
2.5 酸度对捕集的影响
固相捕集剂中的捕集剂TiOA对Np的捕集性能与溶液中的酸度关系很大。Np溶液质量浓度为0.44mg/L,溶液体积5mL,在不同酸度下使用固相捕集剂0.1g制样。计算Np制样回收率(Y)随酸度的变化,结果示于图4。由图4可以看出,酸度提高时,固相捕集剂对Np的回收率增加。通过计算可知,对于Np质量浓度为0.15mg/L的样品溶液,当取液量为4mL、酸度达到5.5mol/L硝酸时,回收率近60%,捕集制得样品中Np质量高于仪器的质量检出下限(0.07μg),可达到测量3EU工艺点样品的要求。

图4 酸度对Np制样回收率的影响Fig.4 Dependence of acid on the recovery rate of Np
2.6 工作曲线的建立
推荐分析流程:测定Np质量浓度为0.14~2.8mg/L的Np样品,取4mL样品,使用氨基磺酸亚铁和浓硝酸将Np调至4价,酸度调至5.5mol/L,使用0.1g固相捕集剂制样,反应时间为10min,测量时间为600s,处理数据时使用Ag-Fe作为内标。
建立工作曲线示于图5。由工作曲线计算得出,使用磁助捕集分离制样时,对Np的质量检测下限为0.12μg,质量浓度检测下限为0.04mg/L,达到测量3EU工艺点样品的要求。

图5 测定Np工作曲线Fig.5 Working curve of analyzing Np
3 结 论
通过使用磁助捕集分离制样法,采用XRF检测溶液中微量Np时,质量浓度检测下限为0.04mg/L,达到了经TEVA萃取色层预分离后3EU样品的检测要求,为测量其他工艺点样品奠定了坚实基础。
[1]杨怀元.液闪计数测定α核素技术的一些进展[J].辐射防护通讯,1997,17(3):15-20.
[2]Rosenberg R J.Non-conventional measurement techniques for the determination of some long-lived radionuclides produced in nuclear fuel a literature survey[J].J Radioanal Nucl Chem,1993,171(2):465-482.
[3]应浙聪.3EU中微量镎的分析方法研究[D].北京:中国原子能科学研究院,2012.
[4]谈炳美,陈耀中,林漳基.氟化镧沉淀载带法测定Np(Ⅲ,Ⅳ)和Np(Ⅴ,Ⅵ)的含率[J].原子能科学技术,1992,26(1):85-89.
[5]全葳.TiOA顺磁性固相萃取剂的研制及分离痕量钚的可行性研究[D].北京:中国原子能科学研究院,2012.
