Parkin基因突变和线粒体功能紊乱与帕金森病的相关性
2015-12-25姜立志,沈丽华,孙翠
Parkin基因突变和线粒体功能紊乱与帕金森病的相关性
姜立志1沈丽华孙翠
(南通大学附属医院神经内科,江苏南通226001)
关键词〔〕帕金森病;Parkin基因;线粒体功能
中图分类号〔〕R742.5〔文献标识码〕A〔
通讯作者:沈丽华(1969-),女,硕士生导师,主任医师,主要从事神经元变形性疾病的临床研究。
据最新流行病学统计,我国65岁以上人群中帕金森病(PD)发病率与西方发达国家相似,约为1.7%,且发病率随着年龄的增加而增加〔1〕。但目前对于PD的确切发病机制尚不明确。研究发现,老年化因素、环境因素、遗传因素均与PD的发病相关。流行病学研究显示,10%~15%的PD患者有家族史〔2〕。家族性PD相关基因见文献〔3〕,家族性PD与基因突变有关,这一发现为疾病的细胞和分子路径提供了一个新的入口〔4〕。迄今为止,已确定的PD相关易感基因有α-synuclein、Parkin、UCH-L1、DJ-1、PINK1、LRRK2、ATP13A2 及HTRA2,其中Parkin、DJ-1、PINK1 与常染色体隐性遗传性PD相关〔5〕。
1Parkin与PD
Parkin最早是Matsumine等〔6〕在研究日本13个常染色体隐性遗传性青少年型PD家系时发现,由Kitada首先克隆并命名〔7〕。Parkin基因的突变在家族性隐性早发性PD中占50%,在早发型散发病例中也占据了15%~20%,发病年龄≤50岁〔8〕。此类患者的特异性临床特点包括:早期出现运动症状伴有缓慢的临床过程、对低剂量的多巴胺敏感、长期治疗可以出现运动障碍的并发症,且常伴有焦虑、精神错乱、强迫症、抑郁〔9〕。Parkin蛋白由3个功能域构成:①氨基端的泛素样区(Ub1),其功能是与底物结合;②羟基端的环指结构,其功能主要是连接泛素结合酶,作为新的锌指蛋白,控制细胞生长、分化与发育,可能与真核细胞分化、成长和死亡控制有关;③前两者的链接区,前两者为功能区,在遗传上高度保守。临床资料显示大片段缺失突变或点突变的突变热点区分别在这两个保守区〔10〕。见图1。
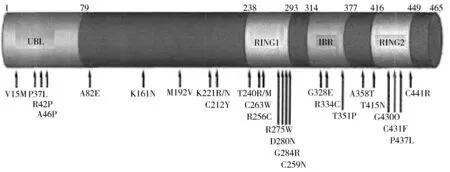
图1 Parkin分子结构及PD相关突变的示意图 〔11〕
1十堰市郧西县人民医院神经内科
第一作者:姜立志(1983-),男,在读硕士,主要从事神经元变形性疾病的临床研究。
Parkin的分布广泛,在脑和外周组织中均有表达。早期的研究报道发现,Parkin在脑的黑质和蓝斑处的神经元有丰富的表达〔12〕。目前研究已证实parkin也可存在于外周组织细胞的胞质、线粒体和内质网中〔13〕。Parkin蛋白具有多重功能,在底物蛋白降解、调节基因转录及保护神经细胞等方面都起着十分广泛的作用,并且在PD的氧化应激、线粒体损伤及蛋白酶体功能紊乱中起着重要作用。
1.1Parkin与泛素蛋白酶体系统(UPS)UPS是细胞内一种重要的蛋白降解途径。Parkin最初是作为E3泛素连接酶被报道,它通过蛋白酶体介导清除细胞中的各种物质〔14〕。细胞内目标蛋白质的特异性降解依赖于泛素分子转运者通过酶的级联反应识别目标蛋白,最终通过UPS降解。因此,UPS功能障碍或者超负荷会导致蛋白质底物的蓄积,在多巴胺神经元中蓄积的蛋白质底物导致多巴胺神经元变性,在脑组织中形成包涵体,称路易小体。Parkin蛋白的底物蛋白已经发现了几十种,如:DJ-1〔15〕、Hsp70〔16〕、PICK1〔17〕、α-酮戊二酸载体蛋白〔18〕等。见图2。
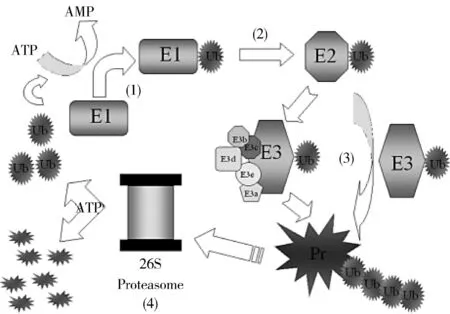
图2 UPS降解底物的示意图 〔19〕
1.2Parkin与线粒体功能线粒体均匀地分布在细胞质中,是物质氧化能量供应的场所,为机体的生命活动提供所需要的能量,同时介导细胞程序化凋亡。PD患者的中脑黑质细胞中,存在一段4 977bp长的DNA缺失,结果导致神经元细胞中线粒体功能缺陷,进而引起神经元能量代谢障碍。因此,线粒体损伤或线粒体功能紊乱是PD发病的主要原因之一,研究发现Parkin与线粒体的功能和形态有着密切的联系。Parkin与线粒体之间的联系最初被观察是在神经酰胺治疗细胞中阻止细胞色素C的释放〔20〕。后来在Parkin基因敲除的动物模型研究中证实Parkin基因在维持线粒体功能中扮演着重要的角色:Parkin敲除鼠出现了线粒体功能紊乱和氧化损伤,Parkin基因剔除果蝇中发现线粒体病变和DA神经元丢失〔20,21〕。同时细胞离体实验也证明了Parkin选择性泛素化凋亡蛋白(Bax),通过阻止线粒体免受Bax的损伤,从而达到抗凋亡作用〔22〕。在线粒体毒素诱导的细胞损伤模型中,Parkin的高表达可以对细胞起到保护作用;而Parkin突变时Bax的表达明显增高,细胞凋亡增加〔22〕。Parkin在线粒体的功能调节中发挥着重要的作用。它通过与线粒体DNA直接结合,保护线粒体DNA免受活性氧(ROS)的损伤并激发线粒体的自我修复过程〔23〕。在增殖的细胞中,Parkin的过表达可以增加线粒体DNA的转录和修复水平,而且Parkin可以通过结合线粒体转录因子A(TFAM)来增加介导的线粒体蛋白转录,从而维持线粒体稳态〔24,25〕。同时线粒体的应激能通过转录激活因子(ATF)4上调诱导特殊ParkinmRNA和蛋白的水平增加〔26〕。
1.3Parkin与线粒体自噬许多研究已证实Parkin在PINK1的参与下通过选择性自噬功能障碍的线粒体,从而参与线粒体的质量控制〔27〕,减少氧化应激,保护细胞免受氧化损伤。自噬是一个动态的过程,它可分为4个阶段:①细胞在饥饿、氧化损伤等情况下,一个杯状膜结构吞噬胞质中的被降解物;②随后这个膜结构逐渐延伸,形成一个囊状结构,也叫自噬体;③自噬体形成后与溶酶体融合,形成自噬溶酶体;④形成的自噬溶酶体允许水解酶降解内部的生物化学部件〔28〕。在正常状态下,Parkin绝大部分定位于胞质中,但有实验证明在线粒体氧化剂羰基氰酯-3-氯苯基腙(CCCP)处理下的细胞中Parkin从胞质迁移到了线粒体,它通过激活自噬系统、调节自噬水平以降解受损的线粒体〔13〕。
2线粒体功能紊乱与PD
线粒体在大多数真核生物的代谢过程中提供能量,调节细胞内钙离子的水平,调控细胞凋亡;清除去极化的自噬体或损伤的线粒体。线粒体有内外两层膜,折叠形成脊;传统上被认为是独立的细胞器,线粒体形成复杂的分枝网是由于它具有移动、融合的活动能力。丰富的线粒体形式在不同的组织中取决于他们对氧化磷酸化的依赖。神经元及心肌、骨骼肌含有较高密度的线粒体,也说明他们特别容易受到线粒体损伤引起的能量缺乏的侵害〔29〕。线粒体稳态的改变在PD患者的组织中已被证实,如细胞培养和基因检测〔30〕。
2.1线粒体功能紊乱与氧化应激氧化应激可被解释为氧化剂和抗氧化剂平衡紊乱时的条件反射,前者是一种潜在的细胞损伤〔31〕。在PD的发病机制中氧化应激起重要作用。线粒体在氧化呼吸的过程中消耗氧气同时传递电子,电子通过氧化呼吸传递给氧生成水。然而线粒体功能紊乱,会使氧不能得到有效利用,导致大量电子外露,直接对氧进行单电子还原,生成氧气离子。氧离子又被位于线粒体基质的超氧化物歧化酶(SOD)歧化生成氧气(O2)和过氧化氢水(H2O2),因此就形成了氧自由基(O-)。氧自由基可以引起生物大分子,如蛋白质、核酸和膜磷脂等损伤,破坏细胞结构,干扰细胞功能和能量代谢〔32〕。在细胞中过多的O-可能导致细胞膜上脂质双分子层的氧化损伤,导致神经元功能破坏和细胞坏死;H2O2可以穿过细胞膜在细胞内发挥它的生物学效应;除此之外金属的氧化还原反应也可成为细胞潜在的毒性〔33〕。
氧化应激使神经元平衡紊乱引起脂质过氧化物的产生,蛋白质改变,DNA突变,同时也引起线粒体通透性转换孔的形成,因此能量供应减低,线粒体动力学异常,干扰线粒体的转运能力,减少神经元适应性,最终导致神经元死亡〔34〕。线粒体通透性转换孔形成的主要后果包括钙快速流出线粒体,减少质子梯度,损伤线粒体呼吸,增加ROS生成,释放凋亡因子到细胞质,线粒体肿胀,线粒体膜破裂〔34〕。
人体细胞中也存在着氧化应激的防御系统,如谷胱甘肽(GSH)、抗氧化剂酶及维生素E(VE)等〔35〕。细胞内高浓度的GSH能拮抗不同的O-,另外,活细胞内各种大量的酶也直接或间接参与O-的清除,如GSH还原酶不仅负责GSH的循环,对氧化物的内源性解毒也起到重要作用〔36〕。疾病早期SOD、GSH过氧化物酶(GSH-Px)升高可能为氧化应激所致。
在生物体内,脑组织更易受氧化反应及其产生的自由基的损害,这是因为:①它含有大量的、高浓度的不饱和脂肪酸;②大脑需要相对更多的O2供应;③脑内自由基清除功能相对缺乏,与肝脏相比,它的GSH、GSH-Px及VE含量较低;④脑组织内铁的含量相对较高。以上几点在黑质中表现得最为突出〔37〕。因此,氧化应激水平的增高更容易损伤多巴胺能神经元。此外,氧化反应的产物影响膜配体与受体的结合〔31〕,如影响多巴胺与多巴胺受体的结合,抑制乙酰胆碱的能力下降。
2.2线粒体功能紊乱与细胞代谢哺乳动物所吸入的O290%进入细胞,在线粒体通过氧化磷酸化为生物体提供80%的ATP,然而20%的氧进入细胞后生成活性氧,他们带有孤电子被称为自由基〔31〕。细胞的氧化呼吸在线粒体中进行,在酶的催化作用下氨基酸、脂肪、糖等供能物质氧化而释放能量,通过电子传递和氧化磷酸化将这些能量以高能磷酸键的形式储存在ATP中,为细胞的各种生命活动提供能量。如在神经元的电子传递中所消耗的能量来源于线粒体〔38〕,也可以说神经元的兴奋性高度依赖线粒体产生的ATP。
2.3线粒体与细胞凋亡有证据证明,PD患者中存在线粒体复合物Ⅰ及氧化还原能力降低,ROS生成增加及氧化应激的发生。上述现象导致线粒体膜电位去极化进而诱导线粒体膜通透性转运孔道开放,大量小分子蛋白物质包括细胞色素C、Bax和凋亡诱导因子等从线粒体膜通透性转运通道释放出来,激活半胱冬肽酶依赖性和非依赖性细胞凋亡的发生〔7〕。同时由于ATP的供应不足,Ga2+内流,细胞内Ga2+浓度增加导致线粒体Na2+、Ga2+质子泵功能下降以及细胞通透性改变,当膜的表面孔道开放时,可释放细胞色素C,诱导凋亡因子分泌最终触发凋亡过程。
3Parkin基因突变与线粒体功能紊乱
Parkin基因的突变具有明显的多样性,该基因有12个外显子,均可有不同程度的序列改变,其中以4、6、7号外显子最为常见,他们都处于突变的热点区域〔39〕。Parkin基因不仅作为泛素连接酶通过UPS参与蛋白质底物的降解,也参与线粒体的功能调节和形态维持〔7〕,同时和线粒体的自噬也有关系。这就带来一个问题,Parkin基因不同位点的突变可能造成不同系统的功能改变,在PD的发病机制中也扮演着不同的角色。
近几年,有关报道认为Parkin2、3外显子的缺失性突变可能是导致线粒体功能紊乱的重要因素。Palacino等〔40〕发现在Parkin3号外显子剔除的小鼠中出现了线粒体氧化应激相关蛋白的减少、线粒体氧化呼吸作用减少和随着年龄增长的氧化损伤增加的现象。Pacelli等〔20〕在2例早发性PD患者的皮肤初级纤维母细胞的研究中发现以前未报道过的复合杂合突变:Parkin基因的2~3号外显子或3号外显子缺失,导致完整的整段蛋白丢失。并观察到几个超微结构的畸形,最主要的是线粒体,认为Parkin基因2、3号外显子可能参与线粒体质量控制系统,这一完整蛋白的丢失与能量代谢损伤有关,解除了对ROS产生的抑制,结果是脂质氧化和过氧化物酶改变。同时他们还认为脂质过氧化反应的产物是PD发病机制中早期标志,甚至在临床表现之前出现,直到异常增高到一定水平才会出现临床症状。Gaweda-Walerych等〔25〕在波兰PD患者中研究Parkin的多态性与线粒体单倍的相互作用时发现:Parkin基因2号外显子的缺失与TFAM和单倍体mtDNA相互关系。
4参考文献
1TianYY,TangCJ,WuJ,et al.Parkinson'sdiseaseinChina〔J〕.NeurolSci,2011;32:23-30.
2ShererTB,BetarbetR,GreenamyreJT.Environment,mitochondria,andParkinson'sdisease〔J〕.Neuroscientist,2002;8(3):192-7.
3ThomasB,BealMF.Parkinson'sdisease〔J〕.HumMolGenet,2007;16(2):183-94.
4LiXT,WangQJ,PanNN,et al.Phosphorylation-dependent14-3-3bindingtoLRRK2isimpairedbycommonmutationsoffamilialParkinson'sdisease〔J〕.PloSOne,2011;6(3):1-13.
5王雪晶,郭纪锋,江泓,等.帕金森相关蛋白Parkin与PINK1的相互作用研究〔J〕.生物化学与生物物理进展,2010;37(9):983-7.
6MatsumineH,SaitoM,ShimodaMS,et al.LocalizationofageneforanautosomalrecessiveformofjuvenileParkinsonismtochromosome6q25.2-27〔J〕.AmJHumGenet,1997;60(3):588-96.
7杨辉,左伋,刘雯,等.Parkin、PINK1、DJ-1 和线粒体功能障碍与帕金森病〔J〕.生命科学,2010;22(10):1009-12.
8WangCD,MaHL,FengXL,et al.Parkindosagemutationsinpatientswithearly-onsetsporadicandfamilialParkinson′sdiseaseinChinese:anindependentpathogenicrole〔J〕.BrainRes,2010;1358:30-8.
9BilgicB,BayramA.DifferentiatingsymptomaticParkinmutationscarriersfrompatientswithidiopathicParkinson′sdisease:contributionofautomatedsegmentationneuroimagingmethod〔J〕.ParkinsRelatDisord,2012;18:562-6.
10GuW,AbbasN,LaguneM,et al.CloningofratparkincDNAanddistributionofparkininratbrain〔J〕.JNeurochem,2000;74:1773-9.
11MandemakersW,MoraisVA,DestrooperB,et al.AcellbiologicalperspectiveonmitochondrialdysfunctioninParkinsondiseaseandotherneurodegenerativedisease〔J〕.JCellSci,2007;120:1707-16.
12DalfoE,PorteroOV,AyalaV,et al.EvidenceofoxidativestressintheneocortexinincidentalLewybodydisease〔J〕.JNeuropatholExpNeurol,2005;64:816-30.
13NarendraD,TanakaA,SuenDF,et al.Parkinisrecruitedselectivelytoimpairedmitochondriaandpromotestheirautophagy〔J〕.JCellBiol,2008;183(5):757-9.
14DuplanE,SevalleJ,ViottiJ,et al.Parkindifferentlyregulatespresenilin-1andpresenilin-2functionsbydirectcontroloftheirpromotertranscription〔J〕.JMolCellBiol,2013;5:132-42.
15OlzmannJA,LiL,ChudaevMV,et al.Parkin-mediatedk63-LinkedpolyubiquitiationtargetsmisfoldedDJ-1toaggresomesviabindingtoHDAC6〔J〕.JCellBiol,2007;178:1025-38.
16MooreDJ,WestAB,DikemanDA.Parkinmediatesthedegradation-independentubiquitinationofHsp70〔J〕.JNeurochem,2008;105:1806-19.
17JochM,AseAR,ChenCX,et al.Parkin-mediatedmonoubiquitinationofthePDZproteinPICK1regulatestheactivityofacid-sensingionchannels〔J〕.MolBiolCell,2007;18(8):3105-18.
18王春喻,曹立,唐北沙,等.Parkin与α-酮戊二酸载体蛋白(OGCP)的相互作用研〔J〕.生物化学与生物物理进展,2010;37(1):49-55.
19HelenC,ArdleyPA.Theroleofubiquitin-proteinligasesinneurodegenerativedisease〔J〕.NeurodegenerativeDis,2004;1:71-87.
20PacelliC,DeRasmoaD,SignorileA,et al.MitochondrialdefectandPGC-1αdysfunctioninparkin-associatedfamilialParkinson′sdisease〔J〕.BiochimBiophysActa,2011;1812:1041-53.
21GreeneJC,WhitworthAJ,AndrewsLA.GeneticandgenomicstudiesofDrosophilaparkinmutantsimplicateoxidativestressandinnateimmuneresponsesinpathogenesis〔J〕.HumMolGenet,2005;14:799-811.
22BethannN,AlisonK,GiuseppeP,et al.TheubiquitinE3ligaseparkinregulatestheproapoptoticfunctionofBax〔J〕.ProcNatlAcadSciUSA,2012;109(16):6283-8.
23RothfussO,FischerH,HasegawaT,et al.ParkinprotectsmitochondrialgenomeintegrityandsupportsmitochondralDNArepair〔J〕.HumMolGenet,2009;18(20):3832-50.
24KurodaY,MitsuiT.Parkinenhancesmitochondrialbiogenesisinproliferatingcell〔J〕.HunMolGenet,2006;15(6):883-95.
25Gaweda-WalerychK,SafranowK,Jasinska-MyqaB,et al.PARK2variabilityinPolishParkinson′sdiseasepatients-interactionwithmitochondrialhaplogroupsq〔J〕.ParkinsonismRelatDisord,2012;18:520-4.
26BoumanL,SchlierfA,LutzAK,et al.ParkinistranscriptionallyregulatedbyATF4:evidenceforaninterconnectionbetweenmitochondrialstressandERstress〔J〕.CellDeathDiffer,2011;18:769-82.
27CaliT,OttoliniD,NegroA,et al.EnhancedparkinlevelsfavorER-mitochondriacrosstalkandguaranteeCa2+transfertosustaincellbioenergetics〔J〕.BiochimBiophysActa,2013;1832:495-508.
28RosaLA,PrzedborskiS.MitophagyandParkinson′sdisease:Beeatentostayhealthy〔J〕.MolCellNeurosci,2013;55:37-43.
29FilostoM,ScarpelliM,CotelliMS.Theroleofmitochondriainneurodegenerativediseases〔J〕.JNeurol,2011;258:1763-74.
30DagdaRK,ChuCT.Mitochondrialqualitycontrol:insightsonhowParkinson’sdiseaserelatedgenesPINK1,parkin,andOmi/HtrA2interacttomaintainmitochondrialhomeostasis〔J〕.JBioenergBiomembr,2009;41:473-9.
31RamalingamM,KimSJ.Reactiveoxygen/nitrogenspeciesandtheirfunctionalcorrelationsinneurodegenerativediseases〔J〕.JNeuralTransm,2012;119:891-910.
32LeeM,KwonBM.EffectsofobovatolonGSHdepletedglia-mediatedneurotoxicityandoxidativedamage〔J〕.NeuroimmPharmacol,2012;7:173-86.
33FilizG,CaragounisA,BicalL,et al.Clioquinolinhibitsperoxide-mediatedtoxicitythroughup-regulationofphosphoinositol-3-kinaseandinhibitionofp53activity〔J〕.IntJBiochemCellBiol,2008;40:1030-42.
34DuH,YanSS.Mitochondrialmedicineforneurodegenerativediseases〔J〕.IntJBiochemCellBiol,2010;42(5):560-72.
35VinishM,AnandA,PrabhakarS.AlteredoxidativestresslevelsinIndianParkinson’sdiseasepatientswithPARK2mutations〔J〕.ActaBiochimPolonica,2011;58(2):165-9.
36BiJ,JiangB.ProtectiveeffectsofcatalpolagainstH2O2-induced oxidative stress in astrocytes primary cultures〔J〕.Neurosci Lett,2008;442:224-7.
37何建成,王振华,袁灿兴,等.复方地黄方对帕金森病大鼠神经行为学及氧化应激的影响〔J〕.中国康复医学杂志,2009;24(7):590-2.
38PatkilG,LauYS.MelatoninprotectsagainstneurobehavioralandmitochondrialdeficitsinachronicmousemodelofParkinson′sdisease〔J〕.PharmacolBiochemBehav,2011;99:704-11.
39刘珂,滕继军,王修海.散发性帕金森病Parkin基因突变检测〔J〕.青岛大学医学院学报,2009;45(2):103-6.
40PalacinoJJ,SagiD,GoldbergMS,et al.MitochondrialdysfunctionandoxdativedamageinParkin-deficientmice〔J〕.JBiolChem,2004;279:18614-22.
〔2013-06-19修回〕
(编辑安冉冉/杜娟)
