以4,4'-联吡啶为配体的木犀草素药物共晶的合成及表征
2015-12-13张羽男殷和美张宇赵巍刘立新张大俊匡海学
张羽男,殷和美,张宇,赵巍,刘立新,张大俊,匡海学
(1.黑龙江中医药大学,哈尔滨 150040;2.黑龙江省药学研究所,佳木斯大学,黑龙江佳木斯 154007)
以4,4'-联吡啶为配体的木犀草素药物共晶的合成及表征
张羽男1,2,殷和美2,张宇2,赵巍2,刘立新2,张大俊2,匡海学1*
(1.黑龙江中医药大学,哈尔滨 150040;2.黑龙江省药学研究所,佳木斯大学,黑龙江佳木斯 154007)
利用溶液挥发法首次合成1个全新的木犀草素-4,4'-联吡啶药物共晶,并通过红外光谱、核磁共振、X射线粉末衍射及差示扫描热量分析等对其表征。选择不同物质量比的木犀草素与4,4'-联吡啶及不同反应溶剂为主要影响因素,探索并确定相应合成条件:木犀草素和4,4'-联吡啶以1∶3的物质量比混合后溶于7.0 mL丙酮溶剂,25℃下搅拌3 h后,于室温下挥发,5 d后得到黄褐色粉末状晶体。结果表明,在极性相对较小的溶剂体系中,反应物分子间形成数目合适且方向饱和的氢键连接,可有效促进药物共晶生成。
木犀草素;4,4'-联吡啶;药物共晶;溶液挥发法
网络出版时间2015-12-25 13:10:48[URL]http://www.cnki.net/kcms/detail/23.1391.S.20151225.1310.034.html
张羽男,殷和美,张宇,等.以4,4'-联吡啶为配体的木犀草素药物共晶的合成及表征[J].东北农业大学学报,2015,46(12):72-78.
Zhang Yunan,Yin Hemei,Zhang Yu,et al.Synthesis and characterization of Pharmaceutical Co-crystal of Luteolin with 4, 4'-Dipyridy[J].Journal of Northeast Agricultural University,2015,46(12):72-78.(in Chinese with English abstract)
药物固体存在形态主要有多晶型、盐、水合物或溶剂化物等。药物研发过程需根据药物性质及目标制剂要求选择药物固体形态。固体形态对药物理化性质影响较大,如溶解度、溶出速率、引湿性及稳定性等。近年来,药物共晶技术作为一种新兴技术在新型药物及其新剂型的研发领域日益受到关注[1]。药物共晶是指药物活性分子(Active pharmaceutical ingredient,API)通过引入共晶形成物(Cocrystal former,CCF),按一定计量比在π-π键和氢键等分子间弱作用力下形成超分子晶体化合物,API和CCF在室温下均为固体[2-4]。药物共晶制备方法按组分形态分为两类:溶液合成法和固体合成法。溶液合成法即合成时API和CCF均为液态,是目前最常用方法,包括溶液挥发法,冷却结晶法等。溶液结晶法的前提是API和CCF在溶液中相互作用力强于API或CCF各自的相互作用力,才可能形成共晶。固态研磨法在机械作用力下使得药物形成共晶,最早关于研磨法制备共晶的报道是1884年WOhler用kügelchen制备出醌和苯二酚的晶体。固态研磨法又分为无液研磨和加液共磨,以研磨过程中是否加入溶剂区分。共晶优势为主分子API可以是酸性分子、碱性分子或非离子化分子,且CCF选择较广泛,包括药用辅料或其他药物。药物形成共晶后药理作用不受影响,但理化性质得到改善,如提高生物利用度、改善稳定性或改变熔点等[5-6],同时对于专利保护的晶型药物,可通过共晶制备新的固体形态,扩大专利保护范围。当前,已有报道药物共晶研究主要集中于CCF选择及其与API空间超分子结合等方面,如可可碱、咖啡因和异烟酰胺作为CCF与槲皮素分别形成的药物共晶,吡拉西坦作为CCF与杨梅素形成共晶等[7-8],而对合成条件系统研究却鲜有报道,无法有针对性地为后续工业化放大生产提供前期参考。因此,如何在已明确API和CCF前提下,系统探索相应药物共晶合成条件极为重要。
木犀草素(Luteolin)是一种天然多羟基黄酮醇类化合物,化学名为3',4',5,7-四羟基黄酮,分子式为C15H10O6。主要存在于菊花、金银花、忍冬花、白毛夏枯草等天然药物,及百里香、芽甘蓝、洋白菜、菜花、甜菜、椰菜和胡萝卜等蔬菜中,以糖苷形式分布于芹菜、青辣椒、紫苏叶等多种植物中。木犀草素纯品为黄色结晶状粉末,作为一种广泛分布于植物界的黄酮类化合物,具有抗菌、抗炎、保护心血管、止咳、祛痰、抗氧化和逆转细菌耐药性等多种药理作用[9-13]。然而,由于木犀草素本身难溶于水,在生物体内生物利用度低且难以成药[14]。将木犀草素制备成药物共晶,将有可能利用难题。目前唯一报道的木犀草素药物共晶是通过异烟酰胺为CCF与木犀草素形成的药物共晶,未系统考虑API和CCF的物质量比或反应溶剂等因素对试验的影响[6]。
本文通过选择不同物质量比的木犀草素与4,4'-联吡啶(4,4'-dipyridy)及不同反应溶剂,利用溶液挥发法首次制备得到木犀草素-4,4'-联吡啶天然药物共晶,确定相应合成条件。以期为木犀草素类天然药物活性成分开发利用提供理论参考。
1 材料与方法
1.1材料
1.1.1仪器
双目解剖体式显微镜(鄂州市贝朗科技有限公司);电子天平FA2004(上海恒平电子天平有限公司);HJ-3数显恒温磁力搅拌器(巩义市予华仪器有限公司);VERTEX 70傅立叶变换红外光谱仪(德国Bruker公司);APEX II CCD单晶衍射仪(德国Bruker公司);D8粉末X-射线衍射仪(德国Bruker公司);AVANCE-III 600MHz型核磁共振仪(德国Bruker公司);HP DSC1差示扫描量热仪(瑞士METTLER TOLEDO公司);KQ-250DE型数控超声波清洗器(昆山市超声仪器有限公司)。
1.1.2药物
木犀草素购于天津市科密欧化学试剂有限公司。
1.1.3试剂
无水乙醇、丙酮、甲醇、4,4'-联吡啶均购于天津市科密欧化学试剂有限公司(AR),异丙醇购于天津市凯通化学试剂有限公司(AR)。
1.2方法
1.2.1探索合成条件
经文献调研[15]和理论分析,选定乙醇和丙酮混合液、乙醇、丙酮、甲醇及异丙醇作反应溶剂,选定木犀草素和4,4'-联吡啶的物质量比为1∶1、1∶2、1∶3、2∶1和1∶4,试验方案见表1。室温下搅拌3 h后,缓慢挥发,得到产品后分析鉴定。
1.2.2试验产物表征
1.2.2.1XRD测试
X-射线粉末衍射是一种定量、定性分析技术,可确定制剂中原料药晶型状态,即晶态、非晶态或二者共存。取等量木犀草素、4,4'-联吡啶以及试验合成样品(约30 mg),研成细粉状,测试条件为:Cu靶,电压30 kV,电量40 mA,扫描速度10°·min-1,测试角度范围在20~50°,室温。

表1 木犀草素和4,4'-联吡啶不同物质的量比及不同溶剂制备表Table 1Preparation table of luteolin and 4,4'-dipyridy in different molar ratios and different solvents
1.2.2.2差热测试
差热法是测定样品与参比物热量差(dQ/dT)与温度关系的技术,通过测定物质熔点进行药物及其制剂鉴别,测定药品纯度,同时区分药物共晶的晶型。取等量木犀草素、4,4'-联吡啶和试验合成样品分别置于坩埚内(约5mg),室温下以10℃·min-1升温至600℃,在氮气保护条件下测试。
1.2.2.3红外测试
红外吸收光谱是根据分子内部原子间相对振动和分子转动等信息确定被测物分子结构的分析方法。红外光谱峰位、峰强及峰形可以判断共晶化合物中可能存在的官能团,可推断分子间可能存在的氢键等分子间作用力。分别取木犀草素、4,4'-联吡啶及合成样品(约10 mg),研细,用KBr压片,相同条件下作红外光谱对照试验,测试波数范围400~4 000 cm-1。
1.2.2.4核磁测试
核磁共振波谱可有效确定药物共晶结构中氢原子和碳原子数目及化学环境,为分析API与CCF分子间作用力提供证据。分别取等量(约5 mg)木犀草素、4,4'-联吡啶以及合成样品溶于DMSO-d6溶剂中,TMS为内标,室温下对以上样品核磁共振谱测定。
2 结果与分析
2.1样品合成
按表1列出的试验方案试验,先量取溶剂置于反应容器内,放置磁力搅拌器上搅拌,并称取定量的木犀草素加入反应容器中,再称取4,4'-联吡啶投置于反应容器内,此时开始计时,在搅拌过程期间,注意避免光照。搅拌3 h后,将反应溶液过滤,留取滤液,放置避光处于室温下缓慢蒸发。试验结果显微照片如图1所示。由图1可见,除C3试验组外,其余试验组产物中均含有淡黄色粉末状和白色条状透明的两种晶体,推测可能是木犀草素晶体和4,4'-联吡啶晶体的混合物。而C3试验组则形成较为规则、均一的黄褐色粉状晶体,推测可能是木犀草素-4,4'-联吡啶的药物共晶。C3试验组合成条件为木犀草素和4,4'-联吡啶的摩尔比1∶3,混合后溶于7.0 mL丙酮溶剂中,室温下搅拌3 h后缓慢挥发,5 d后得到目标产物。
2.2XRD解析
木犀草素与4,4'-联吡啶在不同计量比或不同溶剂条件下形成固体产物、木犀草素与4,4'-联吡啶的物理混合谱图和起始原料的XRD如图2所示。可知,木犀草素、4,4'-联吡啶的物理混合物特征峰位为(2θ):20.4°、23.1°、24.5°、25.1°、26.2°、27.5°、29.7°、31.2°、32.0°、37.7°和39.4°,包含木犀草素纯品谱图和4,4'-联吡啶纯品谱图,其峰位及峰型基本一致。木犀草素、4,4'-联吡啶及两者的物理混合谱图与所有试验组所得固体产物XRD衍射图分别比对,A1特征峰位为(2θ):20.4°、23.1°、24.5°、25.2°、26.3°、27.8°、29.9°、31.4°、32.5°、33.2°、37.8°和39.4°,与木犀草素、4,4'-联吡啶的物理混合物峰位相近,峰型基本相同。所有试验组所得产物的XRD图谱与木犀草素、4,4'-联吡啶的物理混合物XRD谱图对比后发现,只有C3试验组产物谱图呈明显不同,特征峰发生明显改变,其峰位及峰型完全不同。C3试验组产物特征峰位为(2θ):20.5°、22.2°、24.1°、25.3°、27.3°、28.7°、30.4°、31.2°、31.7°、33.9°、37.0°和38.6°,C3试验组产物在22.2°、28.7°、30.4°和33.9°处出现新的特征峰,原本归属为木犀草素的21.2°、25.9°、28.2°和4,4'-联吡啶的22.8°特征峰消失,表明C3试验组生成新的晶相。

图1 试验结果显微照片Fig.1Microscope photos of experimental results
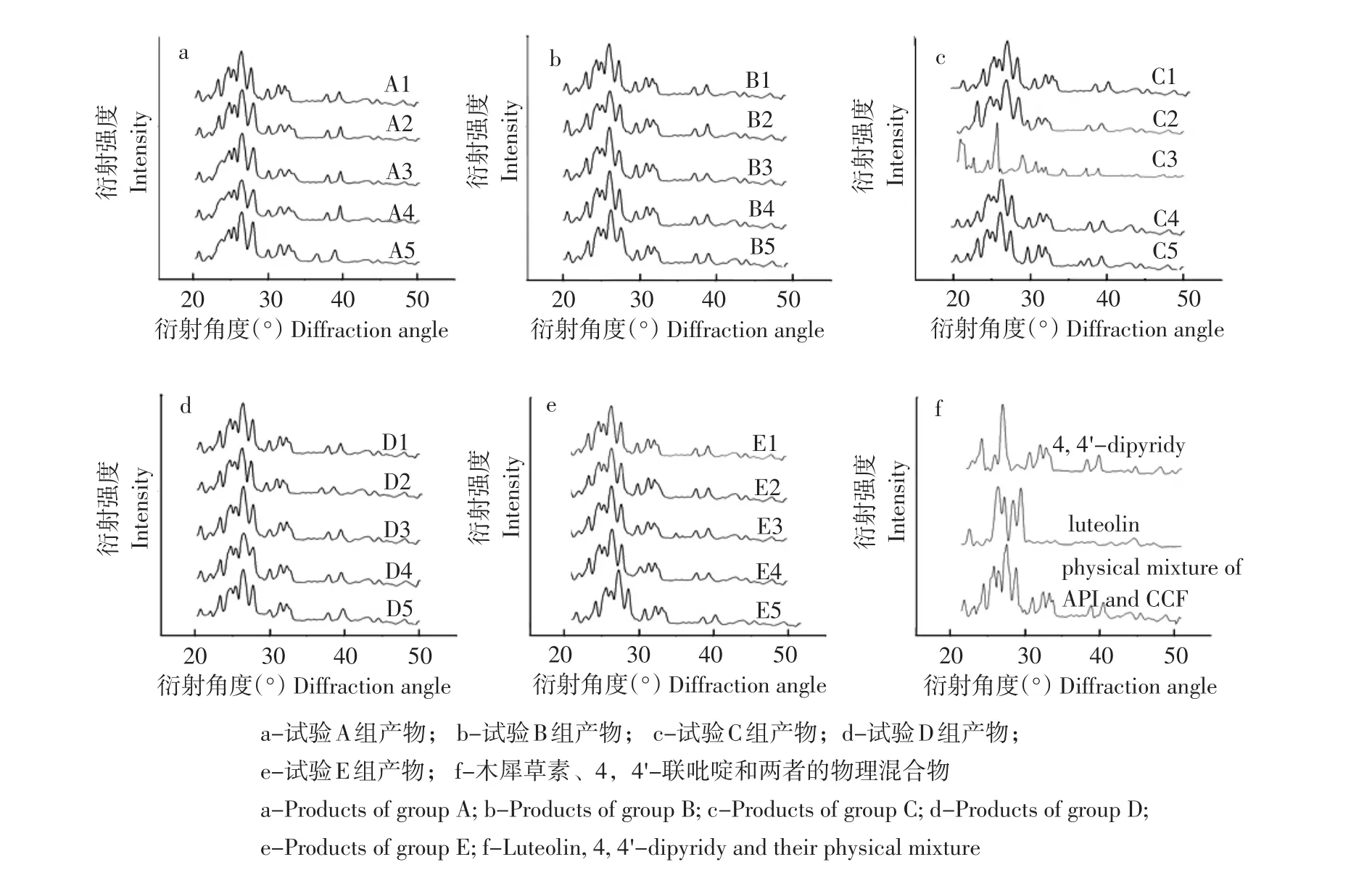
图2 试验结果产物XRD对比Fig.2Comparison of XRD of experimental products
2.3差热解析
C3试验组产物与起始原料的DSC表征结果见图3,由图3可知,木犀草素熔点为341℃,经溶剂挥发法制备的C3试验组产物熔化吸热峰对应温度不同于木犀草素和4,4'-联吡啶,其熔点介于木犀草素和4,4'-联吡啶之间,263和291℃处出现新的吸热峰,原本归属为木犀草素的405℃的峰位及4,4'-联吡啶的227和252℃峰消失,表明有新晶相的形成。C3试验组产物在72、115和340℃的吸热峰与原料药相似,可能是含有未反应完全的原料药所致。

图3 木犀草素、4,4'-联吡啶及C3试验组产物的DSC图Fig.3DSC of luteolin,4,4'-dipyridy and C3 products
2.4红外解析
木犀草素、4,4'-联吡啶和C3试验组产物的红外光谱图如图4所示。C3试验组产物在3 773、3 310、3 214、3 150、2 917、2 834、1 735、1 626、1 585、1 514、1 444、808、732、620和559 cm-1等处有吸收峰,其中位于1 364 cm-1的吸收峰与4,4'-联吡啶的红外光谱ν(-CN)峰相似;位于3 310和1 735 cm-1处吸收峰与山奈酚红外光谱ν(-OH)、ν(-C=O)相似,峰位有所偏移,可能是木犀草素的-OH和4,4'-联吡啶中两端的N原子形成氢键所致;C3试验组产物位于1 585、1 514和1 444 cm-1处吸收峰归属为ν(C=C),位于3 214和3 150 cm-1峰归属为ν(-CH),位于620、732和808 cm-1处吸收峰归属为芳香环上δ(Ar-H)。说明C3试验组产物中含有木犀草素和4,4'-联吡啶。由红外吸收光谱分析可知,木犀草素与4,4'-联吡啶之间可能由于C=N…H-O或C=O…H-O的相互作用,形成共晶化合物。

图4 木犀草素、4,4'-联吡啶和C3试验组产物红外光谱Fig.4IRspectrasofluteolin,4,4'-dipyridyand C3products
2.5核磁解析
如图5所示,C3试验组产物的1H-NMR(400M,DMSO)δC(ppm)12.49(1H,s,5-OH),10.77(1H,s,7-OH),9.57(1H,s,4'-OH),9.44(1H,s,3'-OH),7.68(1H,d,J=2.6 Hz,2'-H),7.51(2H,dd,J=8.5 Hz,2.6 Hz,6'-H),6.87(1H,d,J=8.5 Hz,5'-H),6.39(1H,d,J= 2.4 Hz,8-H),6.17(1H,d,J=2.4 Hz,6-H)和6.07(1H,s,3-H)归属为木犀草素特征峰;8.72(4H,dd,J=5.4 Hz,1.8 Hz)和7.81(4H,dd,J= 5.4 Hz,1.8 Hz)是4,4'-联吡啶上邻位氢和间位氢的化学位移特征信号。

图5 C3试验组产物的1H-NMRFig.51H-NMR of C3 products
C3试验组产物的13C-NMR的δC(ppm)见图6。175.9(C=O)、165.5(C-7)、164.0(C-2)、160.7(C-5)、156.8(C-9)、147.6(C-4')、146.9(C-3')、121.9(C-1')、119.8(C-6')、115.6(C-5')、115.21(C-2')、104.4(C-10)、102.9(C-3)、98.1(C-6)和93.2(C-8)归属为木犀草素特征峰;121.2、144.3和150.5为4,4'-联吡啶芳基碳的特征信号。表明C3试验组产物中存在有木犀草素分子和4,4'-联吡啶分子。

图6 C3试验组产物的13C-NMRFig.613C-NMR of C3 products
3 讨论
药物共晶作为一种改善药物性质的有效手段,具有广泛应用前景,深受研究者青睐。本文在木犀草素的药物共晶制备时选用甲醇、乙醇、丙酮、异丙醇及乙醇与丙酮的混合液5种极性不同的溶剂体系。反应产物经显微镜分析,只有反应溶剂为丙酮时,所得产物不同于其他试验组,形成较为规则均一的黄褐色粉状晶体,而其他4组溶剂作为反应溶剂时得到的均是淡黄色粉末状木犀草素晶体和白色条状透明4,4'-联吡啶晶体的混合物。可能是因为甲醇、乙醇和异丙醇溶剂极性大于丙酮,且均含有-OH,易与木犀草素的-C=O或4,4'-联吡啶两端N原子形成氢键,而不利于木犀草素和4,4'-联吡啶分子间氢键形成导致原料析出,难以形成药物共晶产物。而丙酮溶剂相对于上述其他溶剂,极性较小有利于木犀草素和4,4'-联吡啶分子间氢键形成而促进药物共晶,待后续研究深入分析。
经显微照片分析,C3试验组产物不同于其他试验组产物。对所有试验产物进行XRD测试,除C3试验组产物不同于木犀草素和4,4'-联吡啶物理混合物,其余均与之相似。为进一步验证C3试验组产物为目标产物进行DSC测试,差热分析结果显示,C3试验组产物熔化吸热峰对应的温度不同于木犀草素和4,4'-联吡啶,熔点介于木犀草素和4,4'-联吡啶之间。由红外分析结果可知,木犀草素-4,4'-联吡啶谱图中可见原料药特征吸收峰,几处峰位发生偏移,因木犀草素和4,4'-联吡啶分之间形成氢键,继而形成共晶。
当木犀草素和4,4'-联吡啶的摩尔比为1∶3时合成木犀草素-4,4'-联吡啶的药物共晶。可能是由于1个木犀草素分子含有4个-OH和1个-C=O,一个4,4'-联吡啶分子两端各有1个N原子。药物共晶分子自组装过程中,氢键为主要的分子驱动力与结合力,当木犀草素和4,4'-联吡啶的物质量比为1∶3时,木犀草素分子上的多个-OH和-C=O恰好可与4,4'-联吡啶分子两端的N原子达到数目合适且方向饱和的氢键连接,从而可形成长程有序的药物共晶。
本试验采用溶剂挥发法首次制备得到新木犀草素-4,4'-联吡啶天然药物共晶,确定合成条件为木犀草素和4,4'-联吡啶的物质量比1∶3,混合后溶于7.0 mL的丙酮溶剂,室温下搅拌3 h后缓慢挥发,反应时间为5 d。
4 结论
本文采用溶剂挥发法,以4,4'-联吡啶为共晶配体与木犀草素药物活性分子合成未见报道的新型药物共晶,并进行相应结构表征和性质分析。初步探讨API和CCF物质的量比及反应溶剂等因素对药物共晶制备结果影响,确定相应制备方法和合成条件。理论分析表明,在极性相对较小的溶剂体系中,API和CCF分子间形成数目合适且方向饱和的氢键连接,可有效促进药物共晶生成。为后续木犀草素类天然药物活性成分开发利用及临床前成药研究提供新途径。
[1]Clarke H D,Hickey M B,Moulton B,et al.Crystal engineering of isostructural quaternary multicomponent crystal forms of olanzapine[J].Cryst Growth Des,2012,12(8):4194-4201.
[2]陈学文,宋菊,唐海谊,等.药物共晶筛选与理化性质研究进展[J].中国医药工业杂志,2012,43(8):703-708.
[3]马坤.药物共晶的筛选技术及热力学研究进展[J].药学进展, 2010,34(12):529-534.
[4]易涵,尹亚妹,赵秀丽,等.伊曲康唑共晶的制备与表征[J].沈阳药科大学学报,2014,31(3):161-168.
[5]高缘,祖卉,张建军.药物共晶研究进展[J].化学进展,2010,22 (5):829-836.
[6]Sowa M,slepokura K,Matczak-Jon E.Cocrystals of fisetin, luteolin and genistein with pyridinecarboxamide coformers:crystal structures,analysis of intermolecular interactions,spectral and thermal characterization[J].CrystEngComm.2013,15(38):7696.
[7]Smith A J,Kavuru P,Wojtas L,et al.Cocrystals of quercetin with improved solubility and oral bioavailability[J].Mol Pharmaceut. 2011,8(5):1867-1876.
[8]Sowa M,slepokura K,Matczak-Jon E.A 1:1 pharmaceutical cocrystal of myricetin in combination with uncommon piracetam conformer:X-ray single crystal analysis and mechanochemical synthesis[J].J Mol Struct,2014,1058:114-121.
[9]韩炜,邢燕,康廷国.木犀草素的化学研究进展[J].中国医院药学杂志,2010,30(6):501-503.
[10]肖大凯,覃燕梅,莫丽儿,等.木犀草素对卵巢癌细胞株转移能力的影响[J].中国病理生理杂志,2006,22(6):1199-1202.
[11]张毅,王旭光.木犀草素的体外抗炎机制研究[J].广州中医药大学学报,2007,24(3):231-234.
[12]刘立新,高月林,王朝兴,等.黄酮化合物对金黄葡萄球菌β-内酰胺酶活性影响[J].东北农业大学学报,2013,44(3):119-122.
[13]赵新淮,张强,王竹君.类黄酮化合物抗癌活性研究与进展[J].东北农业大学学报,2010,41(4):133-138
[14]张彬,李敬芬.水溶性木犀草素衍生物的半合成[J].黑龙江医药科学,2008,31(6):72-73.
[15]苏红敏,张婷,罗亚楠,等.两种姜黄素有机药物共晶及其制备方法[P].中国,CN102702092A.2012-10-03.
SynthesisandcharacterizationofPharmaceuticalCo-crystalof Luteolin with 4,4'-Dipyridy
ZHANG Yunan1,YIN Hemei1,ZHANG Yu1,ZHAO Wei1,LIU
Lixin1,ZHANG Dajun1,KUANG Haixue2(1.Institute of Pharmacy in Heilongjiang Province,Jiamusi University,Jiamusi Heilongjiang 154007,China;2.Heilongjiang University of Chinese Medicine, Harbin 150040,China)
A new pharmaceutical co-crystal of Luteolin with 4,4'-dipyridy was firstly synthesized by solution evaporation method.The powder of luteolin-4,4'-dipyridy had been characterized by IR, NMR,XRD and DSC.By selecting different molar ratios of Luteolin and 4,4'-dipyridy and different reaction solvents as the main factors,the preparation condition was to be explored and to be obtained. Luteolin and 4,4'-bipyridy were combined in 7.0 mL acetone with stirring at 25℃for 3 h,and their molar ration was 1∶3.The resulting solution was filtered and allowed to evaporate slowly at room temperature.Then tan powdery crystals were obtained after 5 days.As results,it showed that hydrogen bonds with proper numbers and saturated direction between molecular could promote the formation of pharmaceutical co-crystals in solovent with weak polarity.
luteolin;4,4'-dipyridy;pharmaceutical co-crystal;solution evaporation method
R194
A
1005-9369(2015)12-0072-07
2015-10-17
中国博士后科学基金资助项目(2014M561382);佳木斯大学研究生科技创新项目(LM2015_094);佳木斯大学创新团队项目(CXTD-2013-05)
张羽男(1979-),男,副教授,博士,硕士生导师,研究方向为天然药物共晶的制备。E-mail:zhangyunan79@163.com
匡海学,教授,博士生导师,研究方向为中药药效物质基础。E-mail:hxkuang1954@hotmail.com
