锂离子电池基础科学问题(ⅫⅠ)——电化学测量方法
2015-09-21凌仕刚吴娇杨王少飞
凌仕刚,吴娇杨,张 舒,高 健,王少飞,李 泓
(中国科学院物理研究所,北京 100190)
锂离子电池电极过程一般经历复杂的多步骤电化学反应,并伴随化学反应,电极是非均相多孔粉末电极。为了获得可重现的、能反映材料与电池热力学及动力学特征的信息,需要对锂离子电池电极过程本身有清楚的认识。电池中电极过程一般包括溶液相中离子的传输,电极中离子的传输,电极中电子的传导,电荷转移,双电层或空间电荷层充放电,溶剂、电解质中阴阳离子,气相反应物或产物的吸附脱附,新相成核长大,与电化学反应耦合的化学反应,体积变化,吸放热等过程。这些过程有些同时进行,有些先后发生。
电极过程的驱动力包括电化学势、化学势、浓度梯度、电场梯度、温度梯度。影响电极过程热力学的因素包括理想电极材料的电化学势,受电极材料形貌、结晶度、结晶取向、表面官能团影响的缺陷能,温度等因素。影响电极过程动力学的因素包括电化学与化学反应活化能,极化电流与电势,电极与电解质相电位匹配性,电极材料离子、电子输运特性,参与电化学反应的活性位密度、真实面积,离子扩散距离,电极与电解质浸润程度与接触面积,界面结构与界面副反应,温度等。
为了理解复杂的电极过程,一般电化学测量要结合稳态和暂态方法,通常包括3个基本步骤,如图1所示。
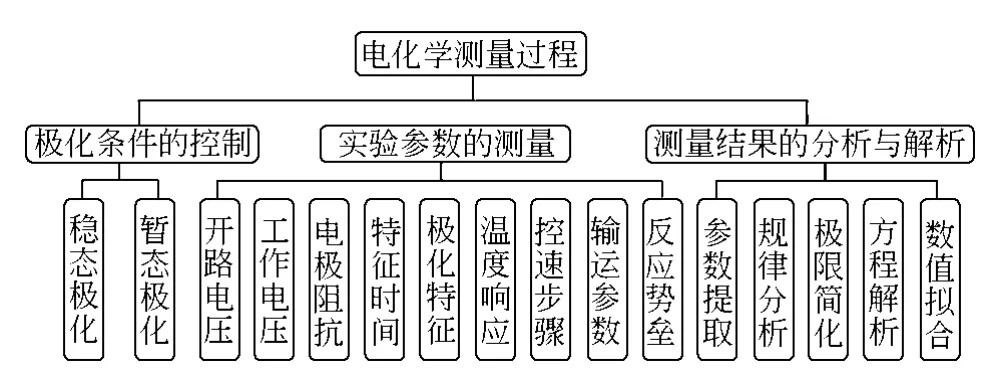
图1 电化学测量的基本步骤Fig.1 Basic steps of electrochemical measurement
1 电化学测量概述[1]
1.1 测量的基本内容
电化学测量主要研究电池或电极的电流、电势在稳态和暂态的激励信号下随外界条件变化的规律,测量反映动力学特性的参数。
1.2 测量电池的分类及特点
电化学测量一般采用两电极电池或三电极电池,较少使用四电极电池。
1.2.1 两电极电池
如图2所示,蓝色虚线框所示是一个典型的两电极电池的测量示意图,其中W表示研究电极,亦称之为工作电极(working electrode),C是辅助电极(auxiliary electrode),亦称之为对电极(counter electrode)。锂电池的研究中多数为两电极电池,两电极电池测量的电压(voltage)是正极电势(potential)与负极电势之差,无法单独获得其中正极或负极的电势及其电极过程动力学信息。
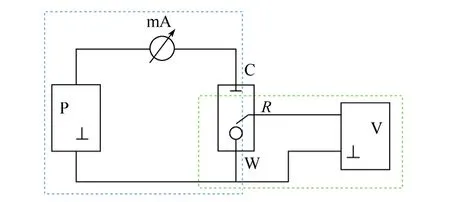
图2 两电极与三电极测量体系示意图Fig.2 Image of two electrode and three electrode system
1.2.2 三电极电池与电极电势以及极化电流的测量
图2是一个三电极电池示意图,W和C分别是工作电极和对电极(同上),R是参比电极(reference electrode)。W和C之间通过极化电流,实现电极的极化。W和R之间通过极小的电流,用于测量工作电极的电势。通过三电极电池,可以专门研究工作电极的电极过程动力学。
由于在锂离子电池中,正极和负极的电化学响应存在较大差异,近年来通过测量两电极电池电压电流曲线,对曲线进行dQ/dV处理,结合熵的原位测量,也能大致判断电池的电流或电压响应主要是与负极还是与正极反应有关。
1.3 参比电极的特性及门类
参比电极的性能直接影响电极电势的准确测量,通常参比电极应具备以下基本特征:①参比电极应为可逆电极;②不易被极化,以保证电极电势比较标准和恒定;③具有较好的恢复特性,不发生严重的滞后现象;④具有较好的稳定性和重现性;⑤快速暂态测量时,要求参比电极具有较低的电阻,以减少干扰,提高测量系统的稳定性;⑥不同的溶液体系,采用相同的参比电极的,其测量结果可能存在差异,误差主要来源于溶液体系间的相互污染和液接界电势的差异。
常用的水溶液体系参比电极有可逆氢电极、甘汞电极、汞-氧化汞电极、汞-硫酸亚汞电极等;常用的非水溶液体系参比电极有银-氯化银电极、Pt电极以及金属锂、钠等电极。此外,也可以用银丝、铂丝做准参比电极,或者采用电化学反应电位稳定的溶解于电解液的二茂铁氧化还原电对。关于准参比电极细节可参考A.J.Bard编著的《Electrochemical Methods》。
1.4 研究电极的门类及特性
电化学测量中常用的研究电极主要有固体电极、超微电极和单晶电极。一般电化学研究所指的的固体电极主要有Pt电极和碳电极。其中碳电极包括热解石墨、高定向热解石墨(HOPG)、多晶石墨、玻璃化碳、碳纤维等。固体电极在使用时需要对其表面进行特殊处理,以期达到较好的重复性。常规的处理步骤为:①浸泡有机溶剂,除去表面吸附有机物;②机械抛光,初步获取较高的表面光洁度;③电化学抛光,除去电极表面氧化层及残留吸附物质;④溶液净化,保证溶液的纯度,消除溶液中的杂质对测量结果的影响。
此外,超微电极和单晶电极以其独特的性质,近些年来也得到了较广泛的应用。前者可以快速获得动力学参数,且对待测材料的量要求很低,可以避免黏结剂、导电添加剂的干扰。后者可以精确获得溶剂吸脱附、表面结构、结晶取向等对电极过程动力学的影响。
在锂离子电池的研究中,固体电极包括含有活性物质的多孔粉末电极、多晶薄膜电极、外延膜薄膜电极、单颗粒微电极以及单晶电极等,多数测量时采用多孔粉末电极。
1.5 电极过程
电极过程一般情况下包括下列基本过程或步骤:①电化学反应过程:在电极/溶液界面上得到或失去电子生成反应产物的过程,即电荷转移过程;②传质过程:反应物向电极表面或内部传递或反应产物自电极内部或表面向溶液中或向电极内部的传递过程(扩散和迁移);③电极界面处靠近电解液一侧的双电层以及靠近电极内一侧的空间电荷层的充放电过程;④溶液中离子的电迁移或电子导体、电极内电子的导电过程。
此外,伴随电化学反应,还有溶剂、阴阳离子、电化学反应产物的吸附/脱附过程,新相生长过程以及其它化学反应等。
锂离子电池作为一种复杂的电化学体系,其电极过程同样具备上述几个基本步骤。其工作原理如图3所示。
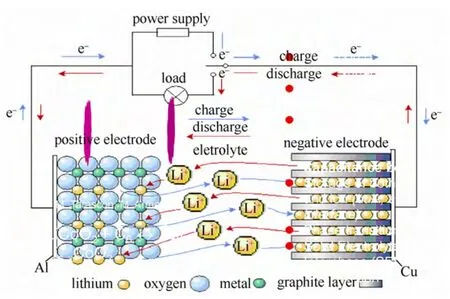
图3 锂离子电池电极过程Fig.3 Electrode processes in Li-ion batteries
针对不同的电极材料及电极体系,上述基本过程可简化为锂离子电池中离子和电子的传输及存储过程。所涉及的电化学过程有电子、离子在材料的体相、两相界面和(solid electrolyte interphase,SEI)的形成等过程。典型的电极过程及动力学参数有:①离子在电解质中的迁移电阻(Rsol);②离子在电极表面的吸附电阻和电容(Rad,Cad);③电化学双电层电容(Cdl);④空间电荷层电容(Csc);⑤离子在电极电解质界面的传输电阻(Rincorporation);⑥离子在表面膜中的输运电阻和电容(Rfilm,Cfilm);⑦电荷转移(Rct);⑧电解质中离子的扩散电阻(Zdiffusion);⑨电极中离子的扩散(Zdiffusion)——体相扩散(Rb)和晶粒晶界中的扩散(Rgb);⑩宿主晶格中外来原子/离子的存储电容(Cchem);相转变反应电容(Cchem);电子的输运(Re)。
上述基本动力学参数涉及不同的电极基本过程,因而具有不同的时间常数。典型的电池中的电极过程及时间常数如图4所示。
图4 储能电池中的动力学过程及典型时间常数Fig.4 Kinetic process in energy storage batteries and its typical time constant
1.6 电化学极化的类型及其特征
1.6.1 极化的类型及其特征
在施加了外来电场后,电池或电极逐渐偏离平衡电势的状态,称之为极化。在不具有流动相的电池中,存在着3种类型的极化:①电化学极化——与电荷转移过程有关的极化,极化的驱动力是电场梯度;②浓差极化——与参与电化学反应的反应物和产物的扩散过程有关的极化,极化的驱动力为浓度梯度;③欧姆极化——与载流子在电池中各相输运有关的极化,驱动力是电场梯度。
若还存在其它基本电极过程,如匀相或多相化学反应过程,则可能存在化学反应极化。
极化电势与平衡电势的差值的大小被称之为过电势。
1.6.2 极化的影响因素
各类极化的影响因素如下。
(1)电化学极化的大小是由电化学反应速率决定的,电化学极化电阻(Rct)的大小与交换电流密度(io)直接相关。受多种因素影响,包括电极电位、电极电位与电解质电化学势差、反应物与产物的活度、参与电化学反应的电极的真实表面积、结晶取向、有序度、表面电导、反应温度、催化剂催化特性、电化学反应的可逆性等。
电化学极化的电流与电势在一定的电流电压范围内一般符合Tafel关系,log(i)与过电势成正比。
(2)浓差极化与传质粒子的扩散系数有关。电池中的扩散过程可以发生在电极材料内部,多孔电极的孔隙中,以及电解质相中,参与扩散的可以是多种带电或中性粒子。涉及扩散的粒子流的流量一般符合菲克扩散定律,与扩散系数及浓度梯度有关。由于电池是非均相体系,扩散系数与浓度梯度是空间位置的函数,在电化学反应的过程中,会随时间变化。传质的快慢与传质距离的平方成正比。
浓差极化过电势ηcon与电流i,极限电流il的关系符合对数关系,ηcon=RT/nF×ln[(il-i)/il]。在过电势较小时,ηcon=-RTi/nFil。
(3)欧姆极化的大小是由电池内部涉及到电迁移的各类电阻之和,即欧姆电阻决定的。欧姆极化过电势与极化电流密度成正比。
2 测量方法[2]
2.1 稳态测量
2.1.1 稳态过程与稳态系统的特征
一个电化学系统,如果在某一时间段内,描述电化学系统的参量,如电极电势、电流密度、界面层中的粒子浓度及界面状态等不发生变化或者变化非常微小,则称这种状态为电化学稳态。
稳态不等同于平衡态,平衡态是稳态的一个特例。同时,绝对的稳态是不存在的,稳态和暂态也是相对的。稳态和暂态的分界线在于某一时间段内电化学系统中各参量的变化是否显著。
2.1.2 稳态极化曲线的测量方法
稳态极化曲线的测量按照控制的自变量可分为控制电流法和控制电势法。
控制电流法亦称之为恒电流法,恒定施加电流测量相应电势。控制电势法亦称之为恒电位法,控制研究电极的电势测量响应电流。
本质上恒电流法和恒电势法在极化曲线的测量方面具有相同的功能,如果电化学体系中存在电流极大值时选择恒电势法,存在电势极大值时选择恒电流法。
2.1.3 稳态测量方法的应用
稳态极化曲线是研究电极过程动力学最基本的方法,在电化学基础研究方面有着广泛的应用。可根据极化曲线判断反应的机理和控制步骤;可以测量体系可能发生的电极反应的最大反应速率;可以测量电化学过程中的动力学参数,如交换电流密度、传递系数、标准速率常数和扩散系数等;可以测定Tafel斜率,推算反应级数,进而获取反应进程信息;此外,还可以利用极化曲线研究多步骤的复杂反应,研究吸附和表面覆盖等过程。
2.2 暂态测量
2.2.1 暂态过程与暂态系统的特征
暂态是相对稳态而言的,随着电极极化条件的改变,电极会从一个稳态向另一个稳态转变,在此期间所经历的不稳定的、电化学参量显著变化的过程称之为暂态过程。
暂态过程具有如下基本特征:①存在暂态电流——该电流由双电层充电电流和电化学反应电流组成,前者又称之为非法拉第电流或电容电流,后者常常称之为法拉第电流;②界面处存在反应物与产物粒子的浓度梯度——即电极/溶液界面处反应物与产物的粒子浓度,如前所述,不仅是空间位置的函数,同时也是时间的函数。
2.2.2 暂态过程中的等效电路分析及其简化
由于暂态过程中的各参量是随时间变化的,与稳态过程比较,更为复杂。为便于分析和讨论,将各电极过程以电路元件组成的等效电路的形式来描述电极过程,等效电路施加电流后的电压响应,应与电极过程的电流电压响应一致。典型的两电极测量体系等效电路如图5所示。
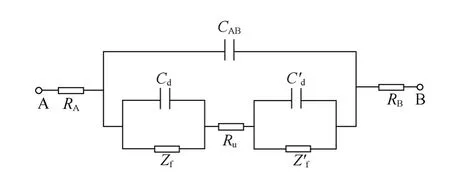
图5 两电极体系电解池的等效电路Fig.5 Equivalent circuit of two electrode system
图5中,A和B分别代表研究电极和辅助电极(两电极体系),RA和RB分别表示研究电极和辅助电极的欧姆电阻,CAB表示两电极之间的电容,Ru表示两电极之间的溶液电阻,Cd和Cd′分别表示研究电极和辅助电极的界面双电层电容,Zr和Zr′分别表示研究电极和辅助电极的法拉第阻抗。
若A、B均为金属电极,则RA和RB很小,可忽略;由于两电极之间的距离远大于界面双电层的厚度,故CAB比双电层电容Cd和Cd′小得多,当溶液电阻Ru不是很大时,由CAB带来的容抗远大于Ru,故CAB支路相当于断路,可忽略;此外,若辅助电极面积远大于研究电极面积,则Cd′远大于Cd,此时,Cd′容抗很小,相当于短路,故等效电路(图5)最终可简化为如图6所示。这相当于在电池中一个电极的电阻很小时的情况,如采用金属锂负极的两电极电池。
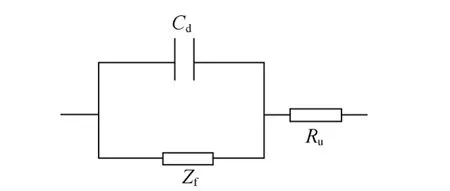
图6 两电极体系电解池的简化电路Fig.6 Simplified circuit of two electrode system
由于电极过程的多步骤和复杂性,不同速率控制步骤下,电极体系的等效电路不尽相同,有时可以进一步简化,常见的有如下三种情形。
(1)传荷过程控制下的等效电路
暂态过程中由于暂态电流的作用使得电极溶液界面处存在双电层充电电流,该双电层类似于平行板电容器,可用Cd表示,相应的充电电流的大小用ic来表示。此外,界面处还存在着电荷的传递过程,电荷的传递过程可用法拉第电流来描述,由于电荷传递过程的迟缓性,导致法拉第电流引起了电化学极化过电势,该电流-电势的关系类似于纯电阻上的电流-电势关系,因而电荷传递过程可以等效为一个纯电阻响应,用Rct表示。由于传荷电阻两端的电压是通过双电层荷电状态的改变而建立起来的,因而,一般认为Rct与Cd在电路中应属于并联关系,传荷过程控制下的简化等效电路如图7所示。需要指出的是,这一简化模型基于传统电化学体系,锂离子电池中,电极在多数状态下。大量电荷存储在电极内,造成电容效应,可以称之为化学电容Cchem,与Cdl应该是串联关系。在实验上与Rct并联显示在阻抗谱半圆上的到底应该是电双层电容还是化学电容还是两种电容之和取决于哪一个电容值更低。
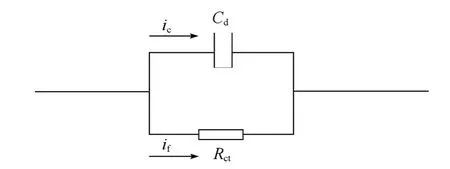
图7 传荷过程控制下的界面等效电路Fig.7 Equivalent circuit of interface under the condition of charge transfer
(2)浓差极化不可忽略时的等效电路
暂态过程中,对于惰性电极,由于电极/溶液界面处存在暂态电流,因此开始有电化学反应的发生,界面处不断发生反应物消耗和产物积累,开始出现反应物产物浓度差。随着反应的进行,浓度差不断增大,扩散传质过程进入对流区,电极进入稳态扩散过程,建立起稳定的浓差极化过电势,由于浓差极化过电势滞后于电流,因此电流-电势之间的关系类似于一个电容响应。可以用一个纯电阻Rw串联电容Cw表示。该串联电路可用半无限扩散模型来模拟,如图8所示。这种情况在电池中也会经常出现。
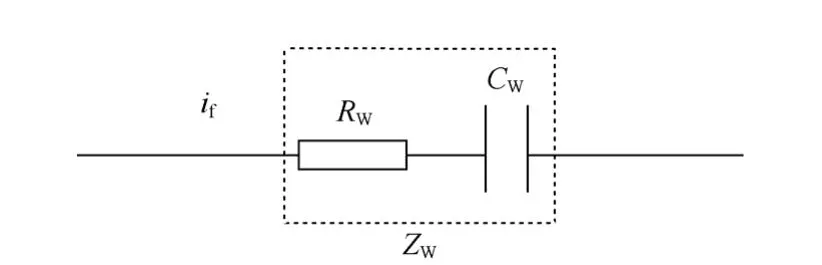
图8 半无限扩散阻抗等效电路Fig.8 Impedance equivalent circuit of semi-infiniti diffusion
上述Rw和Cw的串联结构可用一个复数阻抗Zw来表示,Zw可理解为半无限扩散阻抗。由于扩散传质过程和电荷传递过程同时进行,因而两者具有相同的电化学速率,在电路中应属于串联关系。一般在阻抗谱上表现为45o的斜线。在锂离子电池中,取决于电极材料颗粒尺寸的大小和孔隙率的大小,锂离子在电极材料内部的扩散或者在电极层颗粒之间的孔隙或者含孔颗粒内电解质相的扩散成为控制步骤。由于存在边界条件约束,往往显示出有限边界条件下的扩散。在浓差极化不可忽略的情形下,可以如图9所示。有限边界条件下扩散的等效电路元件只是将Zw换为相应的等效电路扩散元件。
(3)溶液电阻不可忽略时的界面等效电路
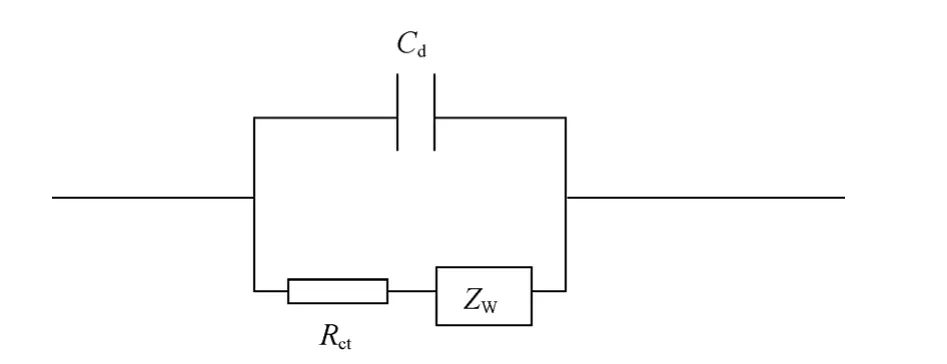
图9 浓差极化不可忽略时的界面等效电路Fig.9 Equivalent circuit of interface under the condition of concentration polarization
当溶液电阻不可忽略时,由于极化电流同时流经界面和溶液,因而溶液电阻与界面电阻应属于串联关系,典型的浓差极化不可忽略、溶液电阻不可忽略时的等效电路如图10所示。在锂离子电池中,由于是多孔粉末电极,有时电极的欧姆电阻也不可忽略,与电解质电阻是串联关系,一般合并在一项中。
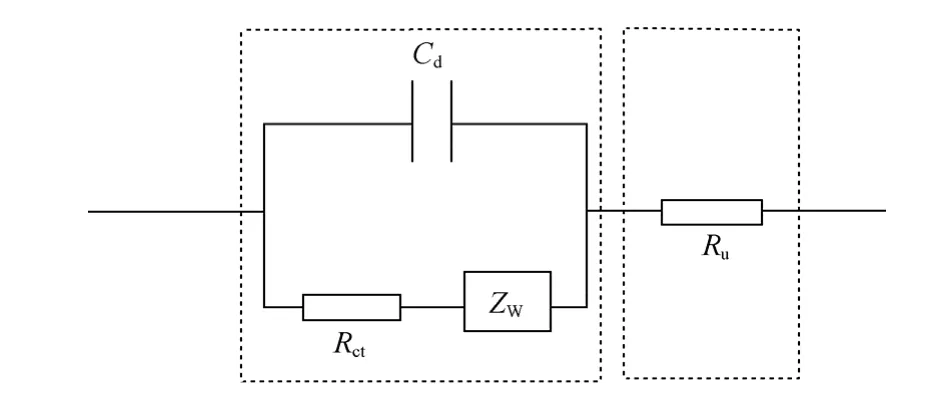
图10 包含4个电极基本过程的等效电路Fig.10 Equivalent circuit including four basic electrode process
2.2.3 暂态测量方法的分类及其特点
暂态过程测量方法按照自变量的控制方式可分为控制电流法和控制电势法;按照自变量的给定方式可分为阶跃法、方波法、线性扫描法和交流阻抗法。用暂态测量能比稳态测量给出更多的电化学参量信息。一般来说,暂态测量法具有如下特点:①暂态法可以同时测量双电层电容Cd和溶液电阻Ru;②暂态法能够测量电荷传递电阻Rct。因此,能够间接测量电化学过程中标准速率常数和交换电流的大小;③暂态法可研究快速电化学反应,通过缩短极化时间,如以旋转圆盘电极代替普通电极,并加快旋转速度,可以降低浓差极化的影响,当测量时间小于10-5s时,暂态电流密度可高达10 A/cm2;④暂态法可用于研究表面快速变化的体系,而在稳态过程中,由于反应产物会不断积累,电极表面在反应时不断受到破坏,因而类似于电沉积和阳极溶解过程,很难用稳态法进行测量;⑤暂态法有利于研究电极表面的吸脱附结构和电极的界面结构,由于暂态测量的时间非常短,液相中的杂质粒子来不及扩散到电极表面,因而暂态法可用于研究电极反应的中间产物和复杂的电极过程。
以上两小节介绍的内容主要适用于传统的电化学体系,氧化还原反应发生在电极表面,电极为惰性电极,电解质为稀浓度电解质,更详细准确的描述参见电化学的教科书。锂电池与传统电化学测量体系显著不同之处是氧化还原反应发生在电极内部而非电极表面,离子的扩散、电荷转移,相变可以发生在电极内部。锂电池的电极一般是非均相多孔粉末电极,孔隙之中存在着电解液,电解液中离子的浓度达到1 mol/L甚至更高,这些不同导致获得可靠的锂离子电池电极过程动力学参数非常困难。而锂空气电池的研究涉及到多种中间产物的分析,圆盘电极和环盘电极等暂态测量被广泛应用。
3 典型的测量方法及其在锂电池中的应用
3.1 锂离子电池电极过程动力学及其测量方法
锂离子电池电极材料在电池充放电过程中一般经历以下几个步骤:①溶剂化的锂离子从电解液内迁移到电解液/固体电极的两相界面;②溶剂化的锂离子吸附在电解液/固体电极的两相界面;③去溶剂化;④电荷转移,电子注入电极材料的导带,吸附态的锂离子从电解液相迁移至活性材料表面晶格;⑤锂离子从活性材料表面晶格向内部扩散或迁移;⑥电子从集流体向活性材料的迁移。
通常,锂离子电池中的电子输运过程比离子扩散迁移过程快很多,锂离子在电解液相中的扩散迁移速度远大于锂离子在固体相中的扩散迁移速度;由于锂离子在固相中的扩散系数很小,一般在10-14~10-9cm2/s数量级,而颗粒尺寸一般在微米量级,因此,锂离子在固体活性材料颗粒中的扩散过程往往成为二次锂电池充放电过程的速率控制步骤。由于电极过程动力学直接关系到电池的充放电倍率、功率密度、内阻、循环性和安全性等性质。对电池与电极过程动力学反应特性的理解以及动力学参数随着充放电过程的演化的定量掌握,对于理解电池中的电化学反应,监控电池的状态,设计电源管理系统具有重要的意义。
锂离子电池的典型工作曲线与测试内容如图11所示。
图11 锂离子电池的典型工作曲线与测试内容Fig.11 Typical working curve and measuring contents of lithium ion batteries
目前,已有多种方法被开发并相继用于锂离子电池电极过程动力学信息的测量,如循环伏安法(CV),电化学阻抗谱(EIS),恒电流间歇滴定技术(GITT),恒电位间歇滴定技术(PITT),电流脉冲弛豫(CPR),电位阶跃计时电流(PSCA),电位弛豫技术(PRT)等。
3.2 稳态测量技术——线性电势扫描伏安法
3.2.1 电势扫描伏安法及其特点
线性电势扫描法通过控制电极电势连续线性变化,同时测量通过电极的响应电流来分析电极过程。
线性电势扫描法可分为单程线性电势扫描法和循环伏安法,对应的电势扫描波形为单程线性电势波和连续三角波。
线性电势扫描法在电化学测量中有着广泛的应用,常用于:①判断电极体系中可能发生的电化学反应;②判断电极过程的可逆性;③判断电极反应的反应物来源;④研究电极活性物质的吸脱附过程。在锂电池的研究中,更多的使用循环伏安法。
3.2.2 循环伏安法及其应用
循环伏安法(cyclic voltammetry)是常见的电化学研究方法之一。在传统电化学中,常用于电极反应的可逆性、电极反应机理(如中间体、相界吸/脱附、新相生成、偶联化学反应的性质等)及电极反应动力学参数(如扩散系数、电极反应速率常数等)的探究。典型的循环伏安过程为:电势向阴极方向扫描时,电活性物质在电极上还原,产生还原峰;向阳极方向扫描,还原产物重新在电极上氧化,产生氧化峰。因而一次扫描,完成一个还原和氧化过程的循环,其电流-电压曲线称为循环伏安曲线。通过循环伏安曲线的氧化峰和还原峰的峰高、对称性、氧化峰与还原峰的距离,中点位置,可判断电活性物质在电极表面反应的可逆程度和极化程度。
由1.5节中的描述可知,电极过程是包括多个步骤的复杂过程。当扩散过程为控制步骤且电极为可逆体系时,采用循环伏安法测量化学扩散系数满足公式(1)[3]
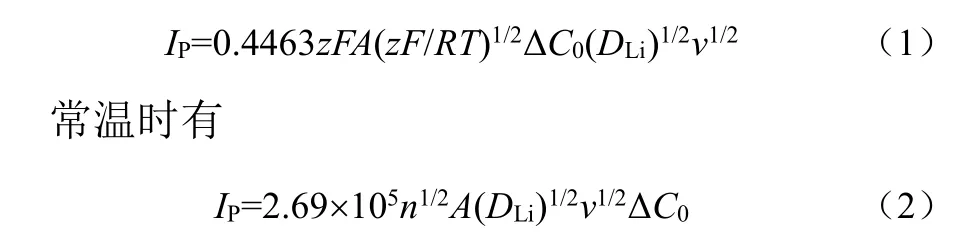
式中,IP为峰电流的大小,n为参与反应的电子数,A为浸入溶液中的电极面积,F为法拉第常数,DLi为Li在电极中的扩散系数,v为扫描速率,ΔC0为反应前后待测浓度的变化。
基本测量过程如下:①测量电极材料在不同扫描速率下的循环伏安曲线;②将不同扫描速率下的峰值电流对扫描速率的平方根作图;③对峰值电流进行积分,测量样品中锂的浓度变化;④将相关参数带入式(2),即可求得扩散系数。不同电极材料中的锂离子扩散系数见表1。

表1 不同电极材料中的锂离子化学扩散系数(CV测量结果)Table 1 Chemical diffusion coefficient of lithium DLiin different electrode material
需要指出的是,上述介绍的循环伏安用于测量化学扩散系数的方法,需要该反应受扩散控制,传统电化学中多用于液相参与反应的物质的扩散。锂离子电池中多数氧化还原反应涉及到固体电极内部的电荷转移,伴随着锂离子嵌入/脱出电极,而这是速率控制步骤,因此多数情况下确实峰电流与扫速的平方根在较宽的扫速范围内满足线性关系,测量的实际是电极内部锂离子与电子的扩散,但也包含了液相锂离子的扩散。此外,对于嵌脱锂引起的连续固溶体反应,化学扩散系数应该是随嵌脱锂量变化的数值,而循环伏安方法计算化学扩散系数时取峰电流值,只能得到表观意义上,在峰值电流对应的反应电位下的平均化学扩散系数。对于两相反应,固体内部不存在连续的浓度梯度,测到的化学扩散系数也是与相转变反应耦合的扩散过程的表观化学扩散系数。可以说循环伏安测到的化学扩散系数并非电极材料内部本征的离子扩散系数。
除了以上影响测量的本征因素,计算化学扩散系数需要知道电极面积。如果是多孔粉末电极,其真实反应面积远大于电极几何面积,且难以精确测量,这给循环伏安方法测量固态电极中化学扩散系数带来了很大的不确定因素,导致数据难以重复,因此根据循环伏安测试结果计算化学扩散系数的绝对值并在不同文献之间比较时需要特别小心。
循环伏安除了可以获得表观化学扩散系数之外,还可以通过一对氧化还原峰的峰值电位差判断充放电(电化学氧化还原反应)之间极化电阻的大小,反应是否可逆。如果氧化与还原反应的过电位差别不大的化,还可以把一对氧化峰与还原峰之间的中点值近似作为该反应的热力学平衡电位值。
恒电流充放电的电压容量曲线微分后以dQ/dV作为纵轴,横轴为电压,可以获得与CV曲线十分相似的结果。因此,在许多文献中,很方便的用V~dQ/dV来代替CV曲线,没有本质区别,因为CV实验中的时间是线性的,电流乘以时间即为电量。
循环伏安方法,通过扫描电势,来观察在某一电势下的电流响应。在扫速非常慢的情况下,在某一电势下发生的电荷转移反应可以进行完全,这一过程获得的CV谱相当于测量一个电极材料的态密度谱,检查在哪个电位下能够注入多少电子。
3.3 准稳态测量技术——交流阻抗谱
3.3.1 交流阻抗法概述
交流阻抗法(alternating current impedance spectroscopy,IS)广泛用于研究介电材料及各类电子元器件,测量时要求待测体系测量端之间没有电压,通过对阻抗谱的分析,获得各类待测元件的阻抗参数。电化学阻抗谱(electrochemical impudence spectroscopy,EIS)是在电化学电池处于平衡状态下(开路状态)或者在某一稳定的直流极化条件下,按照正弦规律施加小幅交流激励信号,研究电化学的交流阻抗随频率的变化关系,称之为频率域阻抗分析方法。也可以固定频率,测量电化学电池的交流阻抗随时间的变化,称之为时间域阻抗分析方法。交流阻抗测量还包括交流伏安法(alternating current voltammetry),是指在线扫伏安的基础上,叠加小幅交流信号,再通过傅里叶变换,研究交流信号的振幅和相位随电势或电流扫描的变化关系。
锂离子电池的基础研究中多用频率域阻抗分析方法,本文只讨论这种测量技术。EIS由于记录了电化学电池不同响应频率的阻抗,而一般测量覆盖了宽的频率范围(μHz~MHz),因此可以分析反应时间常数存在差异的不同的电极过程。EIS数据能够进行有意义的分析要求待测电化学系统满足线性、因果性和稳定性3个基本条件,可以用Kramers-Kronig变换来判断阻抗数据的有效性。
3.3.2 电化学阻抗谱的显示分类
电化学阻抗谱可以有多种展示方法,最常用的为复数阻抗图和阻抗波特图。复数阻抗图是以阻抗的实部为横轴,负的虚部为纵轴绘制的曲线,亦称之为Nyquist图或Cole-cole图。阻抗波特图则由两条曲线组成,其中的一条曲线描述阻抗模量|Z|随频率的变化关系,称之为Bode模量图;另一条曲线描述阻抗的相位角随频率的变换关系,称之为Bode相位图。一般测量时同时给出模量图和相位图,统称为阻抗Bode图。除此之外,还包括介电系数谱(ε′,-ε"),介电模量谱(M′,-M")。
3.3.3 电化学阻抗谱在锂电池基础研究中的应用
当扩散过程为控制步骤且电极为可逆体系时,理想情况下,阻抗低频部分存在扩散响应曲线。此时,可以利用扩散响应曲线测量电池或电极体系的化学扩散系数。典型的采用电化学交流阻抗法测量化学扩散系数的公式见式(3)~(5)[9]
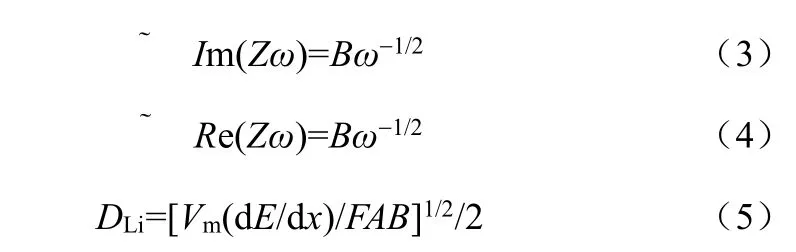
式中,ω为角频率,B为Warburg系数,DLi为Li在电极中的扩散系数,Vm为活性物质的摩尔体积,F为法拉第常数(F=96487 C/mol),A为浸入溶液中参与电化学反应的真实电极面积,dE/dx为相应电极库仑滴定曲线的斜率,即开路电位对电极中Li浓度曲线上某浓度处的斜率。
基本测量过程如下:①通过阻抗谱拟合获得低频扩散部分的B值;②测量库仑滴定曲线;③将相关参数带入方程式(5)即可求出Li的扩散系数。
电化学阻抗谱技术测量不同电极材料中的锂离子化学扩散系数汇总,见表2。
事实上,由于dE/dx很难精确获得,真实面积也存在差异,EIS测到的化学扩散系数的绝对数值往往重现性低,可靠度较差。但是如果是同一个电极,在充放电过程中表面积没有出现大的变化,比较不同充放电状态下同一个电极的扩散系数的变化是合理的。该变化值可以与通过充放电曲线观察过电位的差值变化相对比,从而判断是否电极电阻是扩散控制。
表2中的化学扩散系数通过EIS测量,具有一定的参考价值,有些测量结果还有很大问题,后面的章节会进一步论述。
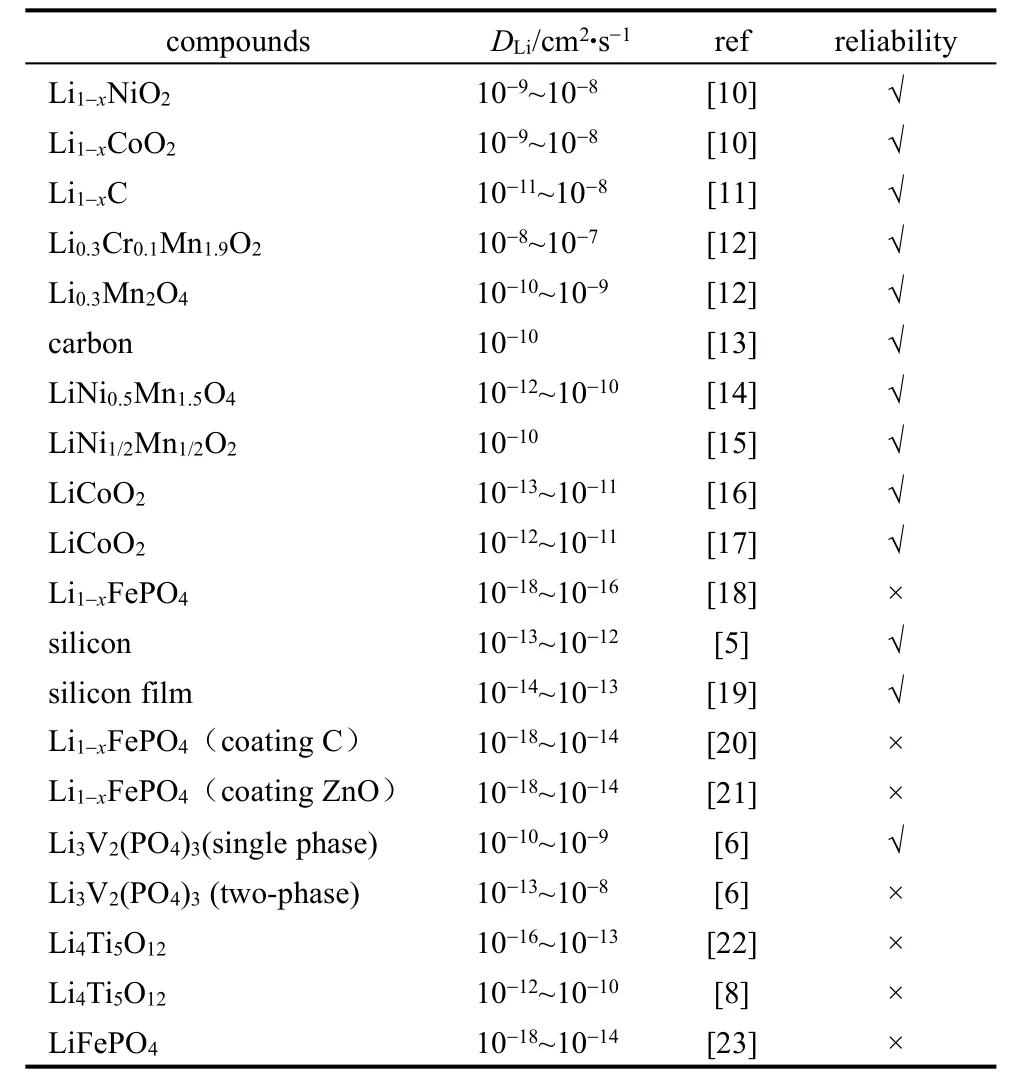
表2 不同电极材料中的锂离子化学扩散系数(EIS测量结果)Table 2 Chemical diffusion coefficient of lithium DLiin different electrode material
3.4 暂态测量技术(Ⅰ)——电流阶跃测量
3.4.1 电流阶跃暂态过程及其特点
电流阶跃暂态测量通过控制流过研究电极的电流,测量电极电势的变化。从而分析电极过程的机理,计算电极过程的有关参数。
对于具有图6所示特性的电化学系统,在某一时刻施加恒定电流[图12(a)],电化学系统的电势响应如图12(b)所示。
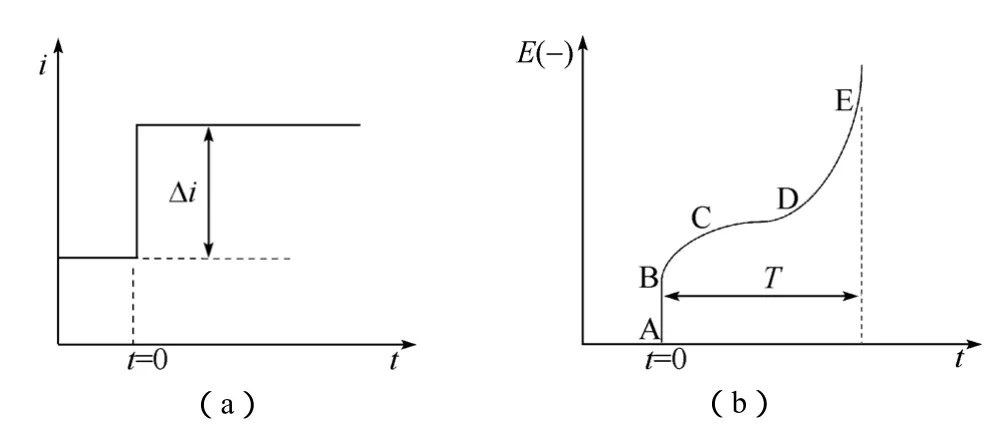
图12 单电流阶跃极化下的控制信号(a)与响应信号(b)Fig.12 Control signal and responded signal under single saltation current
该电极电势随时间响应曲线可做如下分析:AB段为电池欧姆极化过电势,施加恒电流瞬间,界面处双电层电势来不及突变,浓差极化过电势尚未建立,因而表现为欧姆电阻的欧姆极化电势;BC段为电荷传递迟缓性引起的电化学极化过电势,电池中极化电流变化后不久,电化学反应随之开始进行,初期,由于电荷传递的滞后性,导致了BC段的过电势;CD段为浓差极化过电势,随着电化学反应的进一步深入,反应物和反应产物开始出现浓度差。此时浓差极化过电势逐步建立;DE段为双电层充电过电势。当反应持续一段时间后,电极表面反应物浓度逐渐趋于零,浓差极化过电势达到极大值,此时双电层开始快速充电,电极电势发生突变。
上述定性的描述实际是对电池中恒流充放电曲线的一般性描述。而多数电极的充电曲线也确实具有图12(b)的特征。如果AB段极化较大,一般超过10 mV,说明电极电阻较大,这可能与电极材料的电子电导低、导电添加剂电导率低、分散性差以及粘接剂较多、溶剂吸附导致的表面电阻变化有关。BC、CD、DE段的长短和界限对于固溶体类反应很难区分,对于两相反应,BC段往往与颗粒比表面积大小或者材料非计量比范围有关。CD段一般电位恒定,DE段一般斜率较大。
对于同一电极,比较不同电流密度下的充放电曲线,可以获得过电位与电流密度的关系曲线,对于两相反应,由于容易判断充放电平台电位,这一方法较为适用[24-25]。
3.4.2 恒电流间歇滴定技术
恒电流间歇滴定技术(galvanostatic intermittent titration technique)由德国科学家W.Weppner提出,基本原理是在某一特定环境下对测量体系施加一恒定电流并持续一段时间后切断该电流,观察施加电流段体系电位随时间的变化以及弛豫后达到平衡的电压,通过分析电位随时间的变化可以得出电极过程过电位的弛豫信息,进而推测和计算反应动力学信息。
当电极体系满足:①电极体系为等温绝热体系;②电极体系在施加电流时无体积变化与相变;③电极响应完全由离子在电极内部的扩散控制;④τ<<L2/D,L为离子扩散长度;⑤电极材料的电子电导远大于离子电导等条件时,采用恒电流间歇滴定技术测量锂离子化学扩散系数的基本原理可用式(6)表达[26]
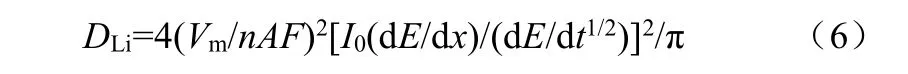
式中,DLi为Li在电极中的化学扩散系数,Vm为活性物质的体积,A为浸入溶液中的真实电极面积,F为法拉第常数(96487 C/mol),n为参与反应的电子数目,I0为滴定电流值,dE/dx为开路电位对电极中Li浓度曲线上某浓度处的斜率(即库仑滴定曲线),dE/dt1/2为极化电压对时间平方根曲线的斜率。
利用GITT方法测量电极材料中的锂化学扩散系数基本过程如下:①在电池充放电过程中的某一时刻,施加微小电流并恒定一段时间后切断;②记录电流切断后的电极电位随时间的变化;③做出极化电压对时间平方根曲线,即dE/dt1/2曲线;④测量库仑滴定曲线,即dE/dx曲线;⑤代入相关参数,利用式(6)求解扩散系数。
不同电极材料中的锂离子化学扩散系数GITT测量结果汇总见表3。

表3 不同电极材料中的锂离子化学扩散系数(GITT测量结果)Table 3 Chemical diffusion coefficient of lithium DLiin different electrode material
固态离子学一般对离子在不同方向上的扩散系数不加区分。对于嵌入化合物而言,从结构的角度考虑,离子嵌入与脱出过程的扩散系数存在着区别。
以正极材料LiNiO2为例,在放电态时,锂离子已占满所有八面体空位LixNiO2(x=1),此时锂离子嵌入时受到Li层已占离子的库仑斥力,而脱出则较为容易。在充电态Li1-xNiO2(x<1),Li层中只有较少的Li+,这时Li+进一步脱出可能会破坏晶体结构,因此受到骨架离子较大的吸引力,而Li+的嵌入则有可能起到稳定结构的作用,此时嵌入阻力小于脱出阻力。
以负极材料石墨为例,Li+的嵌入使石墨层发生膨胀,因此对于初始的石墨材料,Li+的嵌入阻力要大于脱出阻力。当石墨层中已经占满Li+时(LiC6)。Li+的嵌入要克服已占离子的库仑斥力,因此嵌入阻力仍应大于脱出阻力。
因此,根据上述分析,对于正极材料而言,离子嵌入或脱出扩散系数的大小与材料中Li+所占据的量有关,对于负极材料(石墨),锂离子嵌入的扩散系数应小于脱出的扩散系数。
对式(6)分析可以看出,假设嵌入与脱出过程的扩散系数不相等,我们可以通过对处于同一平衡态的电极施加正反向的电流来测得嵌入扩散系数D-(施加负向电流,即放电)与脱出扩散系数D+(施加正向电流,即充电)。
对于同一状态的同一电极,所有参数都一致,因此可以精确比较D+与D-的区别,如式(7)所示[45]
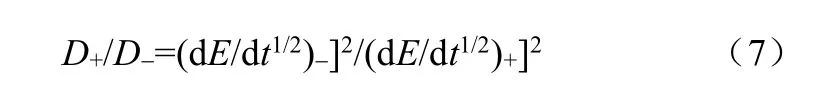
我们对以不同Li/Ni名义组成合成的LixNiO2(x从1.4~0.5)的GITT研究证明[46],D+与D-的比值随x的增加确实出现了交叉变化的现象。在x较大时,D-较小,x较小时D+较大。即在名义组成比n(Li)/n(Ni)>1时,锂离子的嵌入更困难,而n(Li)/n(Ni)<1时,锂离子的脱出更困难。与前述的设想一致。
图13为石墨电极在不同放电深度,LiMn2O4在开路状态时的GITT曲线(这些状态的阻抗谱低频均为扩散控制)。每个电极在同一状态施加不同方向的电流,从图中大致可以看出,放电时电极电位的变化幅度要大于充电时电极电位的变化幅度。图14为LiMn2O4施加电流后E~t1/2的曲线,在测量的范围之内基本为线性,满足GITT的要求。石墨电极E~t1/2曲线也为线性。对图13中所有曲线的计算结果列于表4中。对于石墨电极,其脱出时的扩散系数最大可以超过嵌入时的6倍。LiMn2O4在开路状态离子脱出过程的扩散系数也大于嵌入过程的扩散系数,与设想一致。
在实际锂离子电池中,发现锂离子电池可以快速放电但不能快速充电,充电的过程就是锂离子从正极脱出嵌入碳负极的过程,而锂离子电池的动力学主要受碳材料制约。这种现象与我们用GITT正反电流测试的结果一致,原因正是由于锂离子嵌入碳电极的速率要低于脱出的速率。因此,用这种方法。可以在较短的时间内测定电极材料嵌入脱出扩散系数的差异。结合库仑滴定,可以得到在不同嵌锂量时扩散系数的差异性,从而深入研究结构与动力学之间的关系。
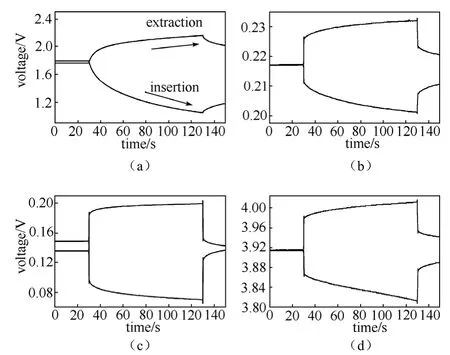
图13 不同三电极电池体系计时电位曲线:(a)天然石墨电极,浅放电态;(b)天然石墨电极,深度放电态;(c)天然石墨,过度放电态;(d)Merck公司锰酸锂产品,开路状态[45]Fig.13 Chronopotentional curves of different electrodes in three-electrode cells:working electrode/1 mol/L LiPF6,EC-DEC(1∶1)/Li/Li.I0=50 μA.(a)natural graphite electrode,a shallow discharged state;(b)natural graphite electrode,a deep discharged state;(c)natural graphite electrode,a deeper discharged state;(d)Merck LiMn2O4electrode,a open-circuit state[45]
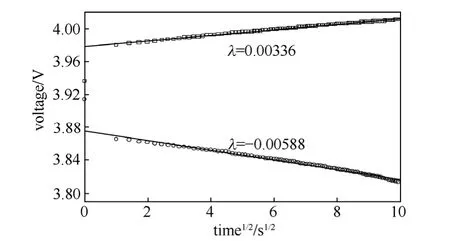
图14 过电位与时间平方根曲线[图13(d)结果][45]Fig.14 A typical E~t1/2curve obtained from Fig.13(d)[45]
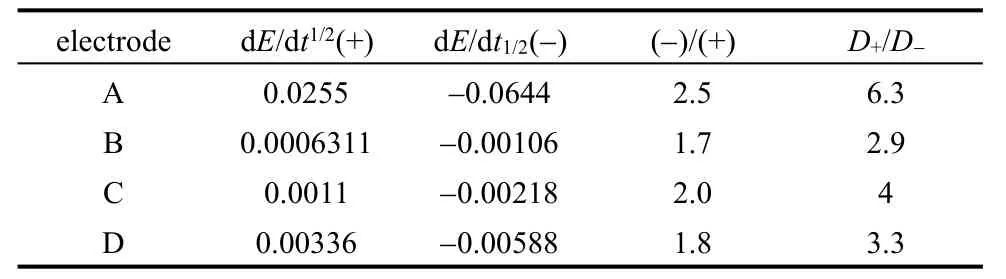
表4 锂离子嵌入与脱出过程中扩散系数的比较(图13计算结果)[45]Table 4 The comparison of the ions insertion/extraction diffusion coefficient.(The values in the table are calculated from the fitting result of Fig.13)[45]
3.5 暂态测量方法 (Ⅱ)——电势阶跃测量
3.5.1 电势阶跃暂态过程及其特点
电势阶跃暂态测量是指控制流过研究电极的电势,按照一定的具有电势突变的规律变化,同时测量电流随时间的变化,从而分析电极过程的机理,计算电极过程的有关参数。
对于具有图6所示特性的电化学系统,在某一时刻施加一恒定电势如图15(a)所示,其电流响应如图15(b)所示。
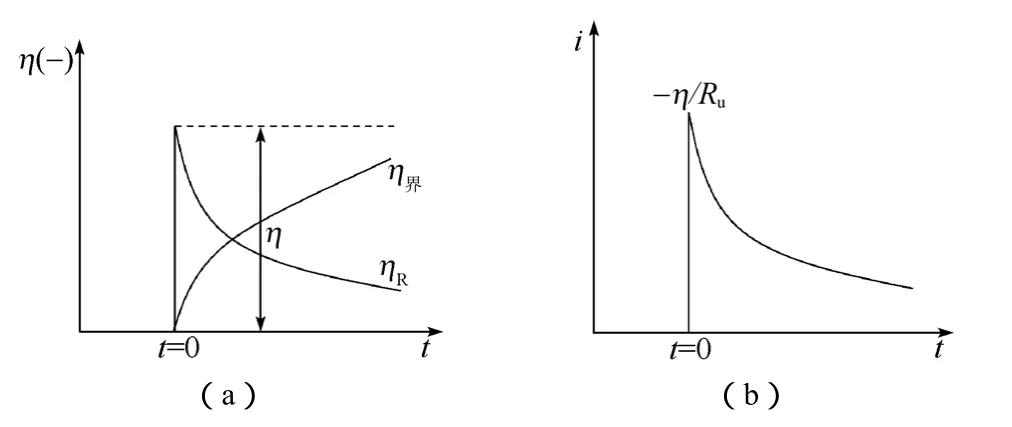
图15 电势阶跃极化下的控制信号(a)与响应信号(b)Fig.15 Control signal(a)and responded signal(b)under single step potential
图15中的控制信号与响应信号之间的关系可依据1.5节中的4个基本过程进行分析:电势阶跃瞬间,由于双电层的作用,电路中的极化电流无法发生突变,极化电流只能慢慢变小,紧随其后的是系统欧姆极化过电势逐步减小,界面双电层过电势逐步增加,两者之和为阶跃电势变化值。
3.5.2 恒电势间歇滴定技术
恒电位间歇滴定技术(potentiostatic intermittent titration technique)是通过瞬时改变电极电位并恒定该电位值,同时记录电流随时间变化的测量方法。通过分析电流随时间的变化可以得出电极过程电位弛豫信息以及其它动力学信息,类似于恒电位阶跃,只是PITT是多电位点测量。
使用恒电位间歇滴定技术测量锂离子化学扩散系数基本原理如下[8]
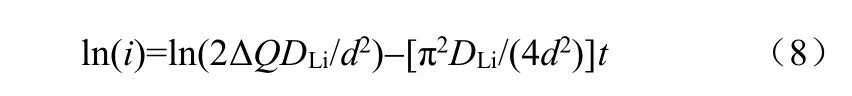
式中,i为电流值,t为时间,ΔQ为嵌入电极的电量,DLi为Li在电极中的扩散系数,d为活性物质的厚度。
基本操作如下:①以恒定电位步长瞬间改变电极电位,记录电流随时间的变化;②利用方程(8)做出ln(i)-t曲线;③截取ln(i)-t曲线线性部分的数据,求斜率即可求出锂离子化学扩散系数。
利用恒电位间歇滴定技术测量不同电极材料中的锂离子化学扩散系数汇总如下,见表5。
对于在充放电过程中动力学明显存在差异的反应体系,电位阶跃能更加突出电流响应的差别,实际上这与循环伏安的结果有类似之处。Chiang等[66]曾用PITT的方法展示磷酸铁锂电极嵌脱锂过程中电流变化更随响应,从而说明其不同段反应的动力学。
表5 不同电极材料中的锂离子化学扩散系数(PITT测量结果)Table 5 Chemical diffusion coefficient of lithium DLiin different electrode material
3.6 暂态测量方法(Ⅲ)—电位弛豫技术
电位弛豫技术(potential relax technique)是在电池与外界无物质和能量交换的条件下研究电极电势随时间的变化关系,该方法属于电流阶跃测量方法中的断电流法,与GITT实验方法一致,不同的是分析弛豫过程中的电位变化。该方法最早由中国科学院物理研究所王庆等[67]运用于锂离子电池电极材料中的离子扩散动力学研究。
采用电位弛豫技术测量锂离子扩散系数的基本原理如式(9)所示[67]
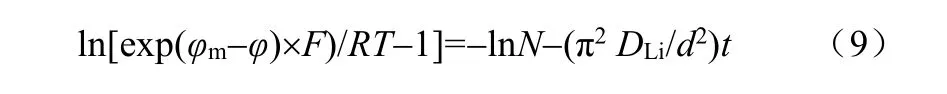
式中,φm为平衡电极电位,φ为初始电位,R为气体常数[8.31 J/(mol·K)],T为绝对温度,d为活性物质的厚度,DLi为Li在电极中的扩散系数,t为电位达到平衡时的时间。
具体测量步骤如下:①对电池预充放电,使电池的库仑效率降至97%左右;②在电池充/放电到一定程度时,切断电流,采用CPT(chrono potentiometry technique)记录电压随时间的变化曲线;③运用式(9)对 ln[exp(φm-φ)×F)/RT-1]-t作图,并对后半部分作线性拟合;④ln[exp(φm-φ)×F)/RT-1]-t曲线进行拟合,求解拟合曲线斜率,带入式(9)即可求得锂的化学扩散系数。
王庆等[67]运用该方法测量了MCMB样品中不同充放电状态下的锂离子化学扩散系数,嵌锂态化学扩散系数数量级在10-8~10-7cm2/s,脱锂态数量级在10-10~10-9cm2/s;与 EIS、GITT、PITT、PSCA、CPR和CV方法不同的是,电位弛豫技术具有较高的精确度,同时可以测量电解质中的锂离子化学扩散系数,具体测量方法可参考文献[67]。吴川等[68]采用该方法测量了氟化物表面修饰的锰酸锂(LiMn2O4)样品中的锂离子化学扩散系数,其化学扩散系数测量值在10-10~10-9cm2/s数量级,并发现了两相界面处的锂离子扩散系数要高于单相区域锂离子的扩散系数。随后,欧阳楚英等[69]又对不同嵌锂量的LixMn2O4(x=0.1~0.9)样品进行了锂离子化学扩散系数的测量,并结合了Monte Carlo方法模拟了锰酸锂样品中的锂离子电导率,证明了在两相界面处(x=0.3~0.7)锂的化学扩散系数要大于单相时的化学扩散系数。
运用电位弛豫技术测量电极过程动力学信息需要满足一定的前提条件。通常,锂离子电池在首周充放电过程中伴随着一些副反应,典型的副反应为SEI膜的形成,为避免副反应的发生对锂离子化学扩散系数测量所带来的干扰,通常电池需要进行几个充放电循环之后开始测量其化学扩散系数。此外,由于电位弛豫是一个非常缓慢的过程,一般在8 h左右,在经过长时间的弛豫后,电位仍不能达到平衡状态,则有可能是仪器漏电所造成,需要特别注意。
3.7 不同电化学测量法的适用范围与精准性
不同的电化学测量方法具有不同的测量精度和适用范围,需要仔细讨论。
唐堃等[23]采用不同的电化学测量方法,系统地研究了LiFePO4薄膜电极中锂的化学扩散系数,并探讨了不同测量方法的适用性和可靠性。
3.7.1 循环伏安法测量LiFePO4薄膜电极中锂离子表观化学扩散系数
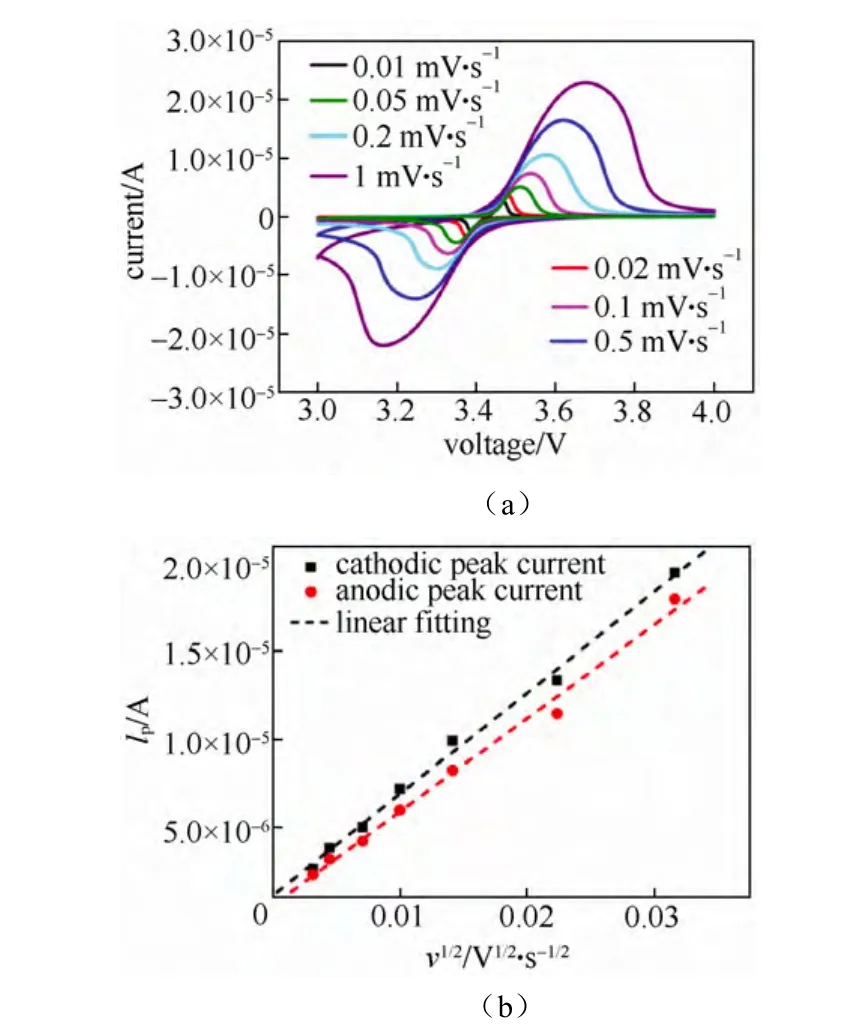
图16 (a)不同电位扫速下的循环伏安曲线;(b)氧化还原峰值电流与电位扫描速率平方根的关系[23]Fig.16 (a)Cyclic voltammograms of the LiFePO4film for various sweep rates and(b)peak current Ip as a function of square root of scan rate v1/2[23]
图16(a)为LiFePO4薄膜电极在不同电位扫描速率下的循环伏安曲线;图16(b)为峰值电流IP对扫描速率平方根v1/2曲线。利用3.2.2节中式(2)的方法计算出的氧化还原过程中锂离子表观化学扩散系数分别为 2.1×10-14cm2/s、1.8×10-14cm2/s。该结果表明,脱锂与嵌锂时的电极过程动力学性质存在微弱差异,该扩散系数应该反映了嵌脱锂在峰值电流附近的平均化学扩散系数,该方法无法得到电极处于不同嵌锂量时的化学扩散系数。
3.7.2 恒电流间歇滴定技术测量LiFePO4薄膜电极化学扩散系数
图17(a)所示为采用GITT法测量LiFePO4薄膜得到的开路电压曲线,图17(b)为利用开路电压曲线拟合得到的库仑滴定曲线。
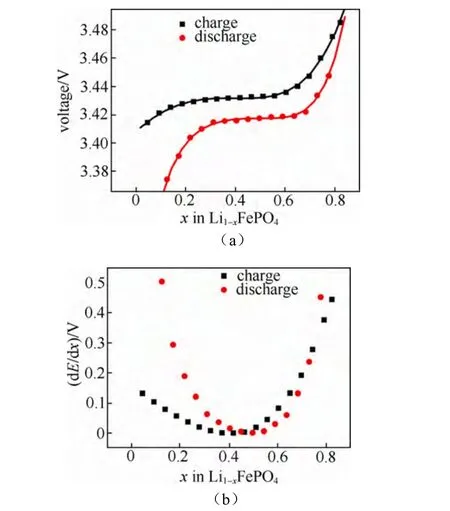
图17 (a)LiFePO4充电和放电开路电压曲线;(b)通过OCV拟合计算所得dE/dx的值[23]Fig.17 (a)The quasi-equilibrium potential vs.Li in Li1−xFePO4as a function of the stoichiometry x;(b)dE/dx as a function of the stoichiometry[23]
图18(a)为电极电势对时间t的平方根曲线,图18(b)为不同锂含量下的dE/dt1/2曲线。
利用3.4.2中式(6)计算出的锂离子化学扩散系数,如图19所示。
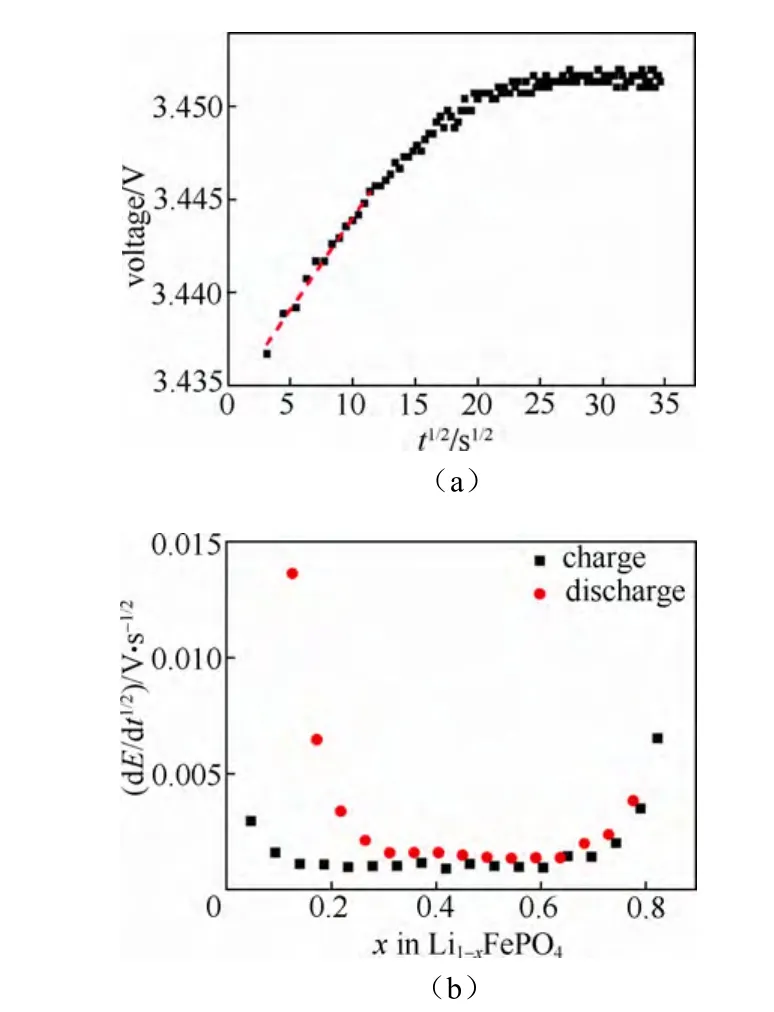
图18 (a)Li0.44FePO4薄膜电极的即时电压与t1/2的关系图;(b)充放电过程中dE/dt1/2与锂离子含量的对应关系图[23]Fig.18 (a)Transient voltage changes as a function of the square root of the time(t1/2)during a single titration process for Li0.44FePO4;(b)dE/dt1/2as a function of the stoichiometry[23]
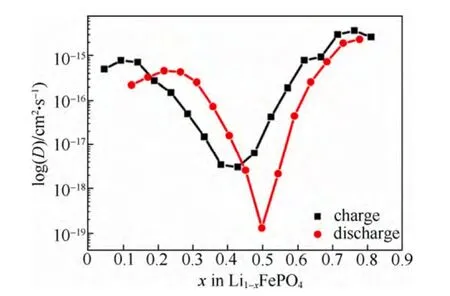
图19 由GITT计算得到LiFePO4的锂化学扩散系数与锂含量的关系[23]Fig.19 Li chemical diffusion coefficients of LiFePO4as a function of stoichiometry from GITT[23]
由图19可以看到锂离子扩散系数在充电过程中处在10-14~10-18cm2/s,随着锂离子的含量变化,化学扩散系数呈V型分布,在x=0.4左右有最小值;而对于放电过程锂的化学扩散系数的分布区间相对较大,在10-14~10-19cm2/s,同样是V型分布,最小值出现在x=0.5左右。这种V型分布的结果,尤其是充电过程最小值在x=0.4处,放电过程最小值在x=0.5处,与图18(b)中的dE/dx值对应。在这两个位置,反应的开路电压基本表现为一条水平线,得到的dE/dx无限接近于零。分析3.4.2节中式(6)可知,对于不同锂含量,体积物质的量浓度Vm、法拉第常数F、施加电流I0、电极表面积A、电极厚度d均为定值,变量只有dE/dx和dE/dt1/2。dE/dt1/2虽然是U型分布,但其随锂含量的变化仍在 0.001~0.01之间,而 dE/dx的数值却在0.001~0.5之间。这说明最终得到的锂离子化学扩散系数的变化规律主要是由于dE/dx的变化导致。而对于两相反应来说,dE/dx在处于平衡态时应为零值,实验记录则不一定为零值,这导致平台区的dE/dx记录没有意义,所以最终得到的锂离子随锂含量的变化规律并不能代表实际的锂离子扩散的快慢。
3.7.3 交流阻抗法测量LiFePO4薄膜中的锂离子化学扩散系数
图20为LiFePO4薄膜电极在不同充电状态的交流阻抗图,图21为阻抗的实部与虚部对ω-1/2的曲线图。利用3.3.3节中式(5)得到的锂离子化学扩散系数如图22所示。
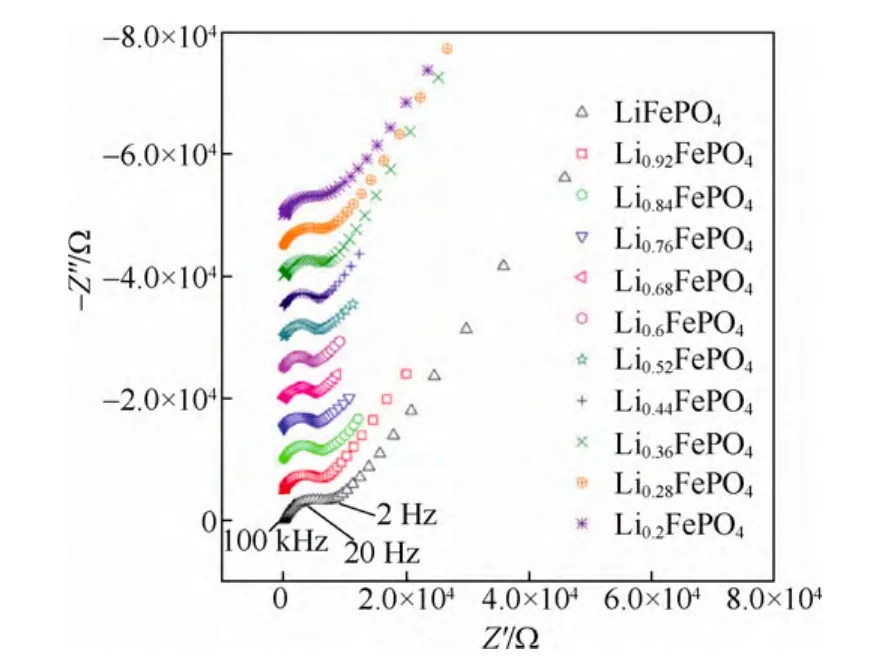
图20 LiFePO4薄膜电极在不同充电状态的交流阻抗图[23]Fig.20 Impedance spectra for Li1-xFePO4at various lithium content x.Frequency range:0.01 Hz to 100 kHz[23]
图22给出了由EIS计算得到的锂化学扩散系数与锂含量的关系图,可以看出,计算得到的锂离子扩散系数的变化规律与由GITT方法测得的变化趋势相同,即在充电的开始和末端有较大的扩散系数,而在中间平台段x=0.5左右有最小的扩散系数。整个区间锂离子扩散系数在10-17~10-13cm2/s之间,数值相差达到四个数量级。分析3.3.3中式(5)可知,整个充电过程中的变量只有Warburg系数B和dE/dx。DLi值与B的平方成反比,与dE/dx值的平方成正比,B的值是随着x的变化呈U型分布,这一数值是可信的。如前所述,对于两相反应,dE/dx会带来相当大的误差,从本质上对两相反应来讲,dE/dx应该始终为零,但是由于测试中的极化问题,使得开路电压曲线只在中间部分表现的接近水平。这样就导致了dE/dx的值在两端和中间相差达到了100倍(0.004~0.445),在经过平方计算以后就相差4个数量级。因此,最终得到的锂化学扩散系数在充电的始末状态和中间状态数值相差四个数量级应该主要归因于dE/dx值的变化,而dE/dx的值对于两相反应是不可信的。
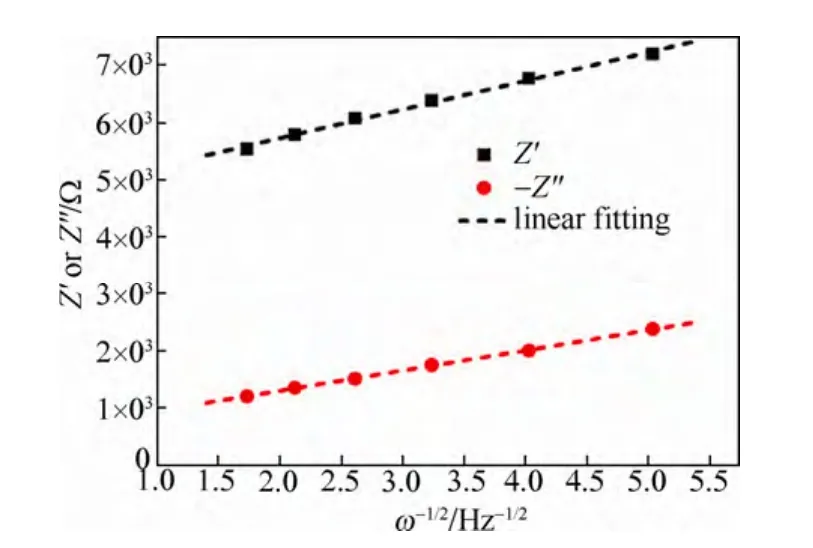
图21 阻抗的实部(Z′)和虚部(Z")对ω-1/2关系图[23]Fig.21 Real and imaginary parts of the complex impedance vs.ω–1/2for Li0.44FePO4[23]
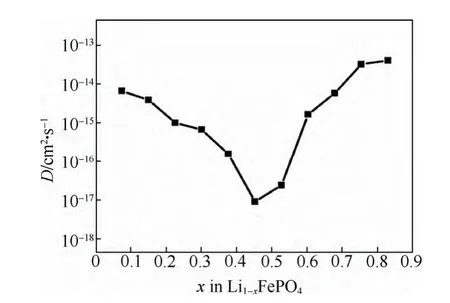
图22 由EIS计算得到的锂离子化学扩散系数与锂含量的关系[23]Fig.22 Li chemical diffusion coefficients as a function of stoichiometry from EIS method[23]
3.7.4 恒电位间歇滴定技术测量LiFePO4薄膜中的锂离子化学扩散系数
图23(a)所示为Li1-xFePO4薄膜电极电位从3.43 V变化到3.435 V,并记录电位恒定过程时的电流随时间变化的曲线,图23(b)为log(i)-t曲线,利用3.5.2节中式(8)计算得到的充放电过程中的锂离子化学扩散系数如图24所示。图25所示为不同锂含量情形下的Li1-xFePO4薄膜电极中锂离子化学扩散系数。
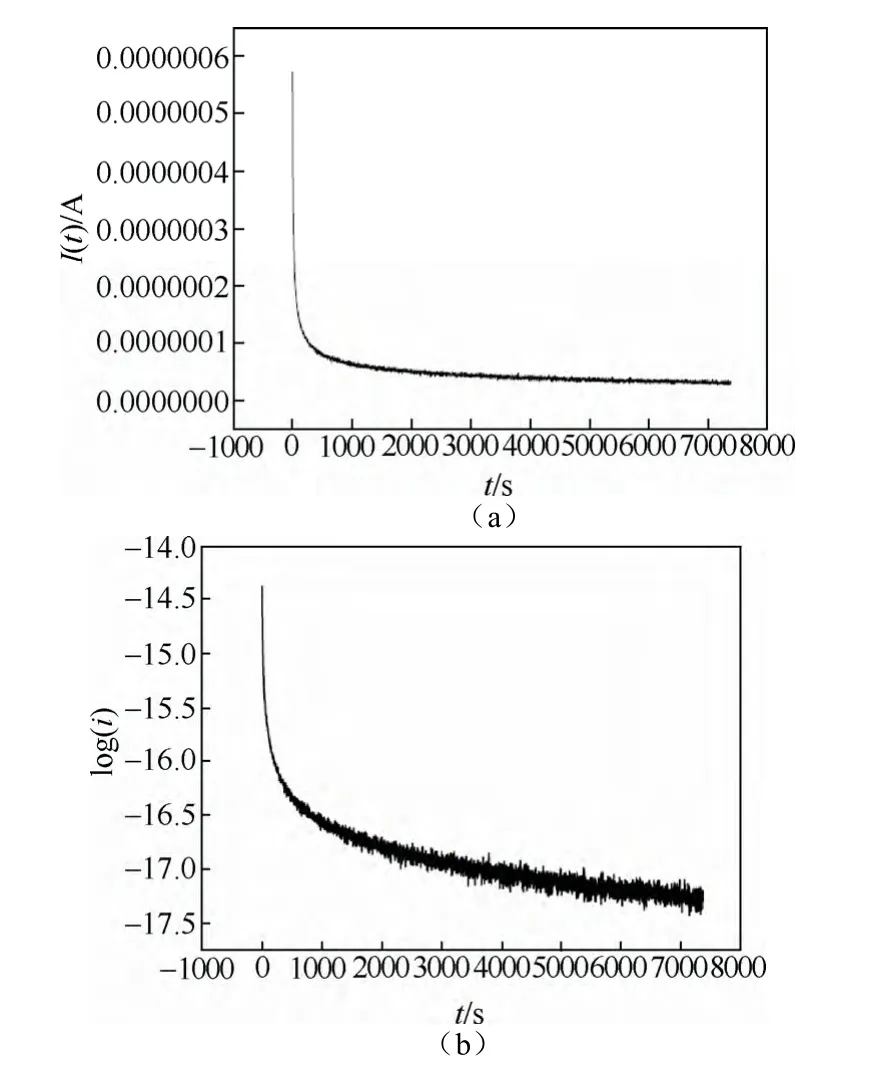
图23 (a)Li1-xFePO4薄膜电极电位从3.43 V变化到3.435 V并恒定过程电位记录的电流随时间变化的曲线;(b)Li1-xFePO4薄膜log(i)vs.t[71]Fig.23 (a)Time dependence of the current in the Li1-xFePO4film for a potential step of 1.44~1.46V vs.Li/Li+:I vs.t(b)log(i)vs.t for Li1-xFePO4thin film[71]
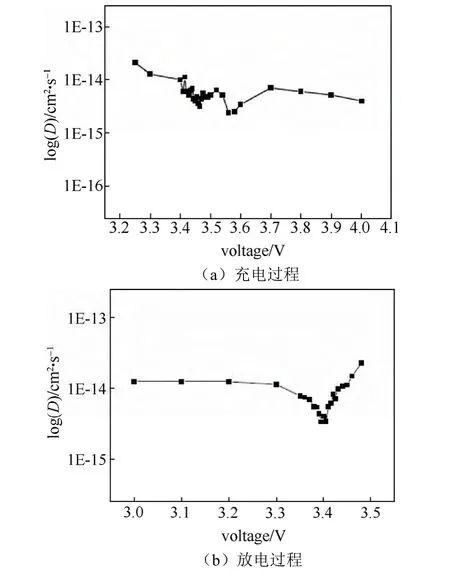
图24 Li1−xFePO4薄膜电极中锂离子扩散系数随电压变化关系(PITT)[71]Fig.24 Relationship between Li chemical diffusion coefficients and its voltage of Li1−xFePO4thin film by PITT(a)charging process and(b)discharging process[71]
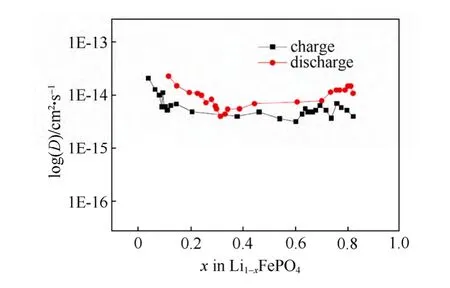
图25 Li1-xFePO4薄膜电极锂离子扩散系数随锂含量的变化(PITT)[71]Fig.25 Relationship between Li chemical diffusion coefficients and lithium contents of Li1-xFePO4thin film by PITT[71]
图24表明充电过程和放电过程锂离子的化学扩散系数在10-15~10-14cm2/s内,变化规律也趋同,即在反应电位附近化学扩散系数有最小值。充电过程和放电过程锂离子的化学扩散系数随锂离子的含量没有明显的变化,对整个区间而言,放电过程的锂离子扩散系数要略高于充电过程的锂离子扩散系数。这一点与3.7.1节中的CV测量结果相悖,但考虑到测试精度,两者相差均在一个数量级范围以内,彼此的比较意义并不是很大。由于计算过程中没有像GITT和EIS方法一样使用到不确切的dE/dx数值,所以,整个锂化学扩散系数的区间处在同一个数量级之间变化。相对而言PITT方法对于LiFePO4这种两相反应的材料锂化学扩散系数的测量的准确性是优于GITT和EIS方法的。而CV方法也应是可靠的,虽然得到的是平均值。
3.7.5 电位弛豫技术测量LiFePO4薄膜中的锂离子化学扩散系数
图26(a)为LiFePO4薄膜样品的电位随时间的弛豫曲线(嵌锂至3.9 V时切断电流),图26(b)为ln[exp(φm-φ)×F)/RT-1]-t曲线,红色虚线为拟合曲线。利用3.6节中式(9)计算得到的锂离子化学扩散系数如图27所示。
利用电位弛豫技术所得锂化学扩散系数随锂离子含量的变化如图27所示。从图中可以看出,利用此方法得到的锂化学扩散系数随着充放电的进行并没有明显的变化,整个区域锂离子的化学扩散系数均在10-15cm2/s左右,整个区间的大小变化在一个数量级以内。充电过程和放电过程的锂离子扩散系数没有明显的区别。且与PITT测量基本一致。由于测量过程中,不涉及dE/dx项的运算,因此,电位弛豫技术对于两相反应类材料的离子化学扩散系数测量是适用的。
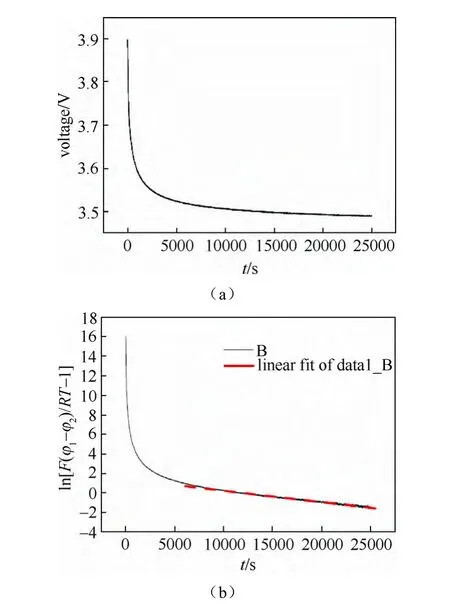
图26 (a)LiFePO4薄膜样品电位随时间的弛豫曲线;(b)ln[exp(φm-φ)×F)/RT-1]-t曲线,红色虚线为拟合曲线[71]Fig.26 (a)Typical potential relaxation profile of graphitized LiFePO4thin film sample(b)Typical curve of ln[exp(φm-φ)×F)/RT-1]-t vs.t.The solid red line indicates the fitting result[71]
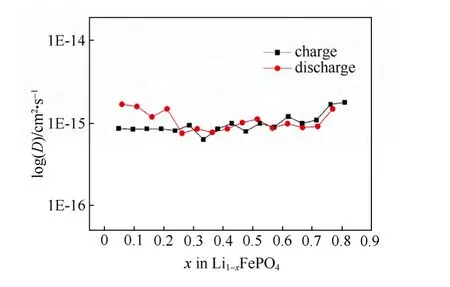
图27 LiFePO4薄膜中锂化学扩散系数(PRT)与锂含量的关系[71]Fig.27 Relationship between Li chemical diffusion coefficients and lithium contents of Li1-xFePO4thin film by PRT[71]
3.7.6 几种测量方法的比较与分析
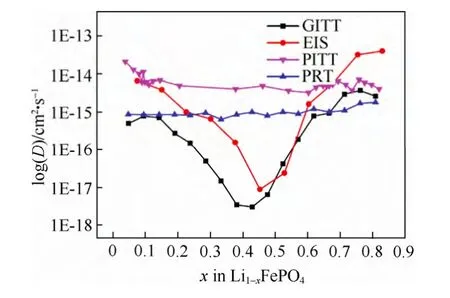
图28 不同测试方法所得到的Li1-xFePO4薄膜中锂离子的化学扩散系数[71]Fig.28 Li chemical diffusion coefficients of Li1-xFePO4thin film by different measuring method[71]
循环伏安方法计算得到LiFePO4薄膜中的表观化学扩散系数为 2.1×10-14~1.8×10-14cm2/s。通过GITT和EIS方法得到的锂化学扩散系数分别在10-14~10-18cm2/s 和 10-14~10-19cm2/s,这两种方法由于计算的过程中均用到了dE/dx,而对于两相反应dE/dx的数值不可能准确测得,且在平台区域其数值近乎为零,所以所得结果均在反应的中段,即平台中部测得了与两端相差四个数量级的最小值,这主要是归因于使用了不准确的dE/dx。计算所得的锂离子扩散系数在反应中段具有最小值这一现象并不能反应真实的物理过程。因此,GITT和EIS方法本身只适用于固溶体体系。相应的,在回避使用dE/dx数值的PITT和PRT方法中,最终的计算结果并没有显示出锂离子扩散系数随锂含量的明显变化规律。PITT方法测得的锂化学扩散系数基本处在10-13~10-14cm2/s之间,而PRT方法测得的锂化学扩散系数则相对要低一些,在10-13cm2/s左右。这两种方法测得的扩散系数在整个区域范围内变化不大,尤其是由PRT计算所得结果基本在同一个数量级上。
以上举例可以看出,电化学测量方法在研究不同反应体系的电极过程动力学时需要慎重考虑。GITT,EIS计算两相反应扩散系数是假设反应时扩散控制,服从Fick扩散定律。而两相反应材料的浓度梯度为一定值,非连续变化,因此数学处理上存在问题。采用Johnson-Mehl-Avrami-Kolmogorov方程来描述相转变动力学更为合适[70]。
3.8 影响电极过程动力学信息测量准确性的基本因素
3.8.1 电极材料对测量结果的影响
对于粉末电极材料,在制备电池的正负极时需要将电极活性物质粉末与黏结剂、导电添加剂混合后形成浆料涂敷在作为集流体的金属箔、网上或者多孔体中。对于这种多孔电极,由于制备时材料的粒度不同,导电添加剂、黏接剂的量及分散程度不同,同时也存在电势的不均匀分布,在测量过程中会引入较多的干扰和不确定性。
相对而言薄膜手段的出现可以在很大程度上消除这些不确定因素。与粉体电极相比,薄膜电极由于直接沉积于金属集流体上,不需要有黏结剂和导电添加剂,所以更加的“干净”,薄膜的形貌可控,可以相对精确地得到其比表面积和离子传输路径(即薄膜厚度);另外薄膜与衬底接触均匀,电势分布相对均匀。最理想的是外延膜电极或单晶电极,制备相对困难。
3.8.2 测量方法对测量结果的影响
对于固溶体即单相反应电极材料而言,在测量体系满足测量模型的前提下,采用循环伏安法、交流阻抗法、恒电流间歇滴定技术、恒电位间歇滴定技术和电位弛豫技术基本可以准确测量电极过程的动力学基本信息,锂电池中最重要的动力学参数为锂离子的化学扩散系数。
对于两相反应,如磷酸铁锂、钛酸锂等,采用GITT、EIS会由于dE/dx无法准确测量而产生较大的误差,循环伏安法由于只能测得表观化学扩散系数,不能测量扩散系数随锂含量的变化规律,有其局限性。因此,对于具体的电极材料和电化学体系,应合理选择测量方法,并构建满足测量方法的测试条件,才能得到真实的测量结果。
3.8.3 测量条件对测量结果的影响
使用EIS、GITT方法测量化学扩散系数,必须保证活性物质的摩尔体积Vm和与溶液的接触面积A恒定,同时需要精确测量库仑滴定曲线,测量结果的可靠性应结合其它测量手段重复定量测试。
此外,测量结果与样品中的锂含量、样品的形貌(单晶、多晶、薄膜)、电极类型(如一维传输,二维平板电极)、充放电状态(充放电截止电压)、电池循环次数、测试温度、相转变反应、样品的修饰(包覆、掺杂)、测试方法、计算参数的获取及测试体系中电解液的选取等都有着很大关系。不同条件下,测得的扩散系数可能存在一到两个数量级的变化。
4 小结与展望
电化学表征技术在锂离子电池中有着非常广泛的应用,而电化学表征方法也非常之丰富,除了文中介绍的几种方法外,还有诸如PSCA、CPR、CITT、RPG等。随着实际应用的需要,新的电化学表征方法,特别是与其它表征技术结合形成的各类原位测量技术,正在迅速发展。
电极过程动力学研究的目的是获得能反映电极材料本征动力学特性的参数值,例如电荷转移电阻、扩散系数、交换电流密度,膜电阻等,并掌握该参数值随不同充放电深度(嵌脱锂量)以及温度的变化,从而能够理解、模拟、预测各类工况下及充电过程中电池极化电阻、电容的变化规律。而实验室在基础研究时往往采用粉末电极,导致在不同材料之间可靠的比较动力学参数基本不可能非常精确,除非材料的尺寸、粒度分布、表面官能团、导电添加剂、粘接剂、分散度、电极厚度、压实密度、体积容量得到了精确的控制和能实现高度的一致性。相对于手工制作的电极,自动化设备制作的电极往往具有较好的一致性,更适合用来研究电极过程动力学。在基础研究时最好采用薄膜电极、微电极或单晶电极。
对于批量生产的电池,通过比较充放电曲线,分析直流极化电阻、固定频率的交流阻抗,开路电压等,可以获得表观的动力学参数,采用这些参数通过电化学模拟软件,可以将为准确的预测电池各类工况下的荷电态、极化电阻、输出功率,成为电源管理系统软件的核心内容[72-73]。
事实上,锂离子电池涉及的电化学为嵌入电极电化学,有别于传统的电极不发生结构演化,电化学反应主要发生在电极表面的溶液电化学。电化学双电层(EDL)与空间电荷层(SCL)共存,在充放电过程中,离子将穿过EDL与SCL,电荷转移往往发生在电极内部而非表面,电极为混合离子导体,电化学反应伴随着相变和内部传质,这与一般教科书上描述的的电化学反应体系、研究方法、数学模型存在显著差异,需要发展新的理论与实验方法[74-78]。
[1]Jia Zheng(贾铮),Dai Changsong(戴长松),ChenLing(陈玲).Electrochemical Measuring Method[M].Beijing:Chemical Industry Press,2006:1-4.
[2]Jia Zheng(贾铮),Dai Changsong(戴长松),Chen Ling(陈玲).Electrochemical Measuring Method[M].Beijing:Chemical Industry Press,2006:46-188.
[3]Das S R,Majumder S B,Katiyar R S.Kinetic analysis of the Li ion intercalation behavior of solution derived nano-crystalline lithium manganate thin films[J].Journal of Power Sources,2005,139:261-268.
[4]Tang S B,Lai M O,Lu L.Li-ion diffusion in highly(003)oriented LiCoO2thin film cathode prepared by pulsed laser deposition[J].Journal of Alloys and Compounds,2008,449(1-2):300-303.
[5]Ding N,Xu J,Yao Y X,et al.Determination of the diffusion coefficient of lithium ions in nano-Si[J].Solid State Ionics,2009,180(2-3):222-225.
[6]Rui X H,Ding N,Liu J,et al..Analysis of the chemical diffusion coefficient of lithium ions in Li3V2(PO4)3cathode material[J].Electrochimica Acta,2010,55(7):2384-2390.
[7]Rui X H,Jin Y,Feng X Y,et al.A comparative study on the low-temperature performance ofLiFePO4/C and Li3V2(PO4)3/C cathodes for lithium-ion batteries[J].Journal of Power Sources,2011,196(4):2109-2114.
[8]Rho Young Ho,Kanamura Kiyoshi.Li ion diffusion in Li4Ti5O12thin film electrode prepared by PVP sol-gel method[J].Journal of Solid State Chemistry,2004,177(6):2094-2100.
[9]Wang C S,Appleby A J,Little F E.Electrochemical impedance study of initial lithium ion intercalation into graphite powders[J].Electrochimica Acta,2001,46(12):1793-1813.
[10]Choi Y M,Pyun S I,Bae J S,et al.Effects of lithium content on the electrochemicallithium intercalation reaction into LiNiO2and LiCoO2electrodes[J].Journal of Power Sources,1995,56(1):25-30.
[11]Levi M D,Aurbach D.Diffusion coefficients of lithium ions during intercalation into graphite derived from the simultaneous measurements and modeling of electrochemical impedance and potentiostatic intermittent titration characteristics of thin graphite electrodes[J].Journal of Physical Chemistry B,1997,101(23):4641-4647.
[12]Tarascon J M,Armand M.Issues and challenges facing rechargeable lithium batteries[J].Nature,2001,414(6861):359-367.
[13]Chang Y C,Sohn H J.Electrochemical impedance analysis for lithium ion intercalation into graphitized carbons[J].Journal of the Electrochemical Society,2000,147(1):50-58.
[14]Mohamedi M,Makino A,Dokko K,et al.Electrochemical investigation of LiNi0.5Mn1.5O4thin film intercalation electrodes[J].Electrochimica Acta,2002,48(1):79-84.
[15]Shaju K M,Rao G V S,Chowdari B V R.Li-ion kinetics and polarization effect on the electrochemical performance of Li(Ni1/2Mn1/2)O2[J].Electrochimica Acta,2004,49(9-10):1565-1576.
[16]Xia H,Lu L,Ceder G.Li diffusion in LiCoO2thin films prepared by pulsed laser deposition[J].Journal of Power Sources,2006,159(2):1422-1427.
[17]Xie J,Imanishi N,Zhang T,et al.Li-ion transport in all-solid-state lithium batteries with LiCoO2using NASICON-type glass ceramic electrolytes[J].Journal of Power Sources,2009,189(1):365-370.
[18]Gao Fei,Tang Zhiyuan.Kinetic behavior of LiFePO4/C cathode material for lithium-ion batteries[J].Electrochimica Acta,2008,53(15):5071-5075.
[19]Xie J,Imanishi N,Zhang T,et al.Li-ion diffusion in amorphous Si films prepared by RF magnetron sputtering:A comparison of using liquid and polymer electrolytes[J].Materials Chemistry and Physics,2010,120(2-3):421-425.
[20]Joachin Humberto,Kaun Thomas D,Zaghib Karim,et al.Electrochemical and thermal studies of carbon-coated LiFePO4cathode[J].Journal of the Electrochemical Society,2009,156(6):A401-A406.
[21]Shenouda Atef Y,Liu Hua K.Studies on electrochemical behaviour of zinc-doped LiFePO4for lithium battery positive electrode[J].Journal of Alloys and Compounds,2009,477(1-2):498-503.
[22]Yuan Tao,Yu Xing,Cai Rui,et al.Synthesis of pristine and carbon-coated Li4Ti5O12and their low-temperature electrochemical performance[J].Journal of Power Sources,2010,195(15):4997-5004.
[23]Tang Kun,Yu Xiqian,Sun Jinpeng,et al.Kinetic analysis on LiFePO4thin films by CV,GITT,and EIS[J].Electrochimica Acta,2011,56(13):4869-4875.
[24]Cui Z H,Guo X X,Li H.Equilibrium voltage and overpotential variation of nonaqueous Li-O2batteries using the galvanostatic intermittent titration technique[J].Energy Environ.Sci.,2015,8:182-187.
[25]Kaifu Zhong,Xin Xia,Bin Zhang,et al.MnO powder as anode active materials for lithium ion batteries[J].Journal of Power Sources,2010,195(10):3300-3308.
[26]Bae J S,Pyun S I.Electrochemical lithium intercalation reaction of anodic vanadium-oxide film[J].Journal of Alloys and Compounds,1995,217(1):52-58.
[27]Shaju K M,Rao G V S,Chowdari B V R.Electrochemical kinetic studies of Li-ion in O2-structured Li2/3(Ni1/3Mn2/3)O2and Li(2/3)+x(Ni1/3Mn2/3)O2by EIS and GITT[J]. Journal of the Electrochemical Society,2003,150(1):A1-A13.
[28]Shaju K M,Rao G V S,Chowdari B V R.EIS and GITT studies on oxide cathodes,O2-Li(2/3)+x(Co0.15Mn0.85)O2(x=0 and 1/3)[J].Electrochimica Acta,2003,48(18):2691-2703.
[29]Shaju K M,Rao G V S,Chowdari B V R.Li ion kinetic studies on spinel cathodes,Li(M1/6Mn11/6)O4(M=Mn,Co,Co,Al)by GITT and EIS[J].Journal of Materials Chemistry,2003,13(1):106-113.
[30]Montoro L A,Rosolen J M.The role of structural and electronic alterations on the lithium diffusion in LixCo0.5Ni0.5O2[J].Electrochimica Acta,2004,49(19):3243-3249.
[31]Shaju K M,Rao G V S,Chowdari B V R.Influence of Li-ion kinetics in the cathodic performance of layered Li(Ni1/3Co1/3Mn1/3)O2[J].Journal of the Electrochemical Society,2004,151(9):A1324-A1332.
[32]Schwenzel J,Thangadurai V,Weppner W.Developments of high-voltage all-solid-state thin-film lithium ion batteries[J].Journal of Power Sources,2006,154(1):232-238.
[33]Levi M D,Aurbach D.The application of electroanalytical methods to the analysis of phase transitions during intercalation of ions into electrodes[J].Journal of Solid State Electrochemistry,2007,11(8):1031-1042.
[34]Ito Atsushi,Li Decheng,Lee Yunsung,et al.Influence of Co substitution for Ni and Mn on the structural and electrochemical characteristics of LiNi0.5Mn1.5O4[J].Journal of Power Sources,2008,185(2):1429-1433.
[35]Wang X J,Yu X Q,Li H,et al.Li-storage in LiFe1/4Mn1/4Co1/4Ni1/4PO4solid solution[J].Electrochemistry Communications,2008,10(9):1347-1350.
[36]Xie J,Kohno K,Matsumura T,et al.Li-ion diffusion kinetics in LiMn2O4thin films prepared by pulsed laser deposition[J].Electrochimica Acta,2008,54(2):376-381.
[37]Xia Hui,Lu Li,Lai M O.Li diffusion in LiNi0.5Mn0.5O2thin film electrodes prepared by pulsed laser deposition[J].Electrochimica Acta,2009,54(25):5986-5991.
[38]Li Zhe,Du Fei,Bie Xiaofei,et al.Electrochemical Kinetics of the Li Li0.23Co0.3Mn0.47O2Cathode Material Studied by GITT and EIS[J].Journal of Physical Chemistry C,2010,114(51):22751-22757.
[39]Zhu Yujie,Wang Chunsheng.Galvanostatic intermittent titration technique for phase-transformation electrodes[J].Journal of Physical Chemistry C,2010,114(6):2830-2841.
[40]Delacourt C,Ati M,Tarascon J M.Measurement of lithium diffusion coefficient in LiyFeSO4F[J].Journal of the Electrochemical Society,2011,158(6):A741-A749.
[41]Rui X H,Yesibolati N,Li S R,et al.Determination of the chemical diffusion coefficient of Li+in intercalation-type Li3V2(PO4)3anode material[J].Solid State Ionics,2011,187(1):58-63.
[42]Wunde F,Berkemeier F,Schmitz G.Lithium diffusion in sputter-deposited Li4Ti5O12thin films[J].Journal of Power Sources,2012,215:109-115.
[43]Yang Shunyi,Wang Xianyou,Yang Xiukang,et al.Determination of the chemical diffusion coefficient of lithium ions in spherical Li Ni0.5Mn0.3Co0.2O2[J].Electrochimica Acta,2012,66:88-93.
[44]Xiao P F,Lai M O,Lu L.Transport and electrochemical properties of highpotential tavorite LiVPO4F[J].Solid StateIonics,2013,242:10-19.
[45]Li Hong(李泓).锂离子电池负极材料及电极过程研究[D].Beijing:Institute of Physics Chinese Academy of Sciences,1999.
[46]Lu Zhonghua(鲁中华).锂离子电池富锂层状正极材料的制备和电化学性能研究[D].Beijing:Institute of Physics Chinese Academy of Sciences,1996.
[47]Dell′Era A ,Pasquali M.Comparison between different ways to determine diffusion coefficient and by solving Fick′s equation for spherical coordinates[J].Journal of Solid State Electrochemistry,2009,13(6):849-859.
[48]Fell Christopher R,Carroll Kyler J,Chi Miaofang,et al.Synthesis-structure-property relations in layered,"Li-excess"oxides electrode materials Li Li(1/3-2x/3)NixMn(2/3-x/3)O2(x=1/3,1/4,and 1/5)[J].Journal of the Electrochemical Society,2010,157(11):A1202-A1211.
[49]Meethong Nonglak,Kao Yu-Hua,Carter W Craig,et al.Comparative study of lithium transport kinetics in olivine cathodes for Li-ion batteries[J].Chemistry of Materials,2010,22(3):1088-1097.
[50]Xie J,Imanishi N,Zhang T,et al.Electrochemical performance of all-solid-state Li batteries based LiMn0.5Ni0.5O2cathode and NASICON-type electrolyte[J].Journal of Power Sources,2010,195(24):8341-8346.
[51]Manjunatha H,Venkatesha T V,Suresh G S.Kinetics of electrochemical insertion of lithium ion into LiFePO4from aqueous 2 M Li2SO4solution studied by potentiostatic intermittent titration technique[J].Electrochimica Acta,2011,58:247-257.
[52]West W C,Soler J,Smart M C,et al.Electrochemical behavior of layered solid solution Li2MnO3-LiMO2(M=Ni,Mn,Co) Li-ion cathodes with and without alumina coatings[J].Journal of the Electrochemical Society,2011,158(8):A883-A889.
[53]Churikov Alexei V,Romanova Veronica O.An electrochemical study on the substituted spinel LiMn1.95Cr0.05O4[J].Ionics,2012,18(9):837-844.
[54]Liu Li,Zhou Meng,Yi Lanhua,et al.Excellent cycle performance of Co-doped FeF3/C nanocomposite cathode material for lithium-ion batteries[J].Journal of Materials Chemistry,2012,22(34):17539-17550.
[55]Li Juchuan,Yang Fuqian,Xiao Xingcheng,et al.Potentiostatic intermittent titration technique(PITT)for spherical particles with finite interfacial kinetics[J].Electrochimica Acta,2012,75:56-61.
[56]Manjunatha H,Mahesh K C,Suresh G S,et al.Kinetics of lithium insertion into LiMnPO4from aqueous saturated LiOH:A study using galvanostatic and potentiostatic intermittent titration techniques[J].Electrochimica Acta,2012,80:269-281.
[57]Rho Y H,Kanamura K.Li ion diffusion in Li4Ti5O12thin film electrode prepared by PVP sol-gel method[J].Journal of Solid State Chemistry,2004,177(6):2094-2100.
[58]Wu Feng,Wang Feng,Wu Chuan,et al.Rate performance of Li3V2(PO4)3/C cathode material and its Liion intercalation behavior[J].Journal of Alloys and Compounds,2012,513:236-241.
[59]Amalraj Francis,Talianker Michael,Markovsky Boris,et al.Study of the lithium-rich integrated compound xLi2MnO3·(1-x)LiMO2(x≈0.5;M=Mn,Ni,Co;2:2:1)and its electrochemical activity as positive electrode in lithium cells[J].Journal of the Electrochemical Society,2013,160(2):A324-A337.
[60]Baster Dominika,Zheng Kun,Zajac Wojciech,et al.Toward elucidation of delithiation mechanism of zinc-substituted LiFePO4[J].Electrochimica Acta,2013,92:79-86.
[61]Lee Jungbae,Kumar Purushottam,Lee Jinhyung,et al.ZnO incorporated LiFePO4for high rate electrochemical performance in lithium ion rechargeable batteries[J].JournalofAlloys and Compounds,2013,550:536-544.
[62]Xiao Liang,Guo Yonglin,Qu Deyu,et al.Influence of particle sizes and morphologies on the electrochemical performances of spinel LiMn2O4cathode materials[J].Journal of Power Sources,2013,225:286-292.
[63]Dai Kehua,Mao Jing,Li Zitao,et al.Microsized single-crystal spinel LAMO forhigh-powerlithium ion batteriessynthesized via polyvinylpyrrolidone combustion method[J].JournalofPower Sources,2014,248:22-27.
[64]Lepage D,Sobha F,Kuss C,et al.Delithiation kinetics study of carbon coated and carbon free LiFePO4[J].Journal of Power Sources,2014,256:61-65.
[65]Li Zheng,Ban Chunmei,Chernova Natasha A,et al.Towards understanding the rate capability of layered transition metal oxides LiNiyMnyCo1-2yO2[J].Journal of Power Sources,2014,268:106-112.
[66]Meethong Nonglak,Huang Hsiao-Ying Shadow,Speakman Scott A,etal.Strain accommodation during phase transformationsin olivine-based cathodes as a materials selection criterion for high-power rechargeable batteries[J]. Advanced Functional Materials,2007,17(7):1115-1123.
[67]Wang Qing,Li Hong,Huang Xuejie,Chen Liquan.Determination of chemical diffusion coefficient of lithium ion in graphitized mesocarbon microbeadswith potentialrelaxation technique[J].Journal of the Electrochemical Society,2001,148(7):A737-A741.
[68]Wu C,Wu F,Chen L Q,et al.Fabrications and electrochemical properties of fluorine-modified spinel LiMn2O4for lithium ion batteries[J].Solid State Ionics,2002,152:327-334.
[69]Ouyang C Y,Shi S Q,Wang Z X,et al.Experimental and theoretical studies on dynamic properties of Li ions in LixMn2O4[J].Solid State Communications,2004,130(7):501-506.
[70]Yu Xiqian,Wang Qi,Zhou Yongning,et al.High rate delithiation behaviour of LiFePO4studied by quick X-ray absorption spectroscopy[J].Chemical Communications,2012,48(94)11537-11539.
[71]Tang Kun(唐堃).磷酸盐正极薄膜材料的制备和电化学性能研究[D].Beijing:Institute of Physics Chinese Academy of Sciences,2009.
[72]Santhanagopalan Shriram,Guo Qingzhi,White Ralph E.Parameter estimation and model discrimination for a lithium-ion cell[J].Journal of the Electrochemical Society,2007,154(3):A198-A206.
[73]Schmidt Alexander P,Bitzer Matthias,Imre Arpad W,et al.Experiment-driven electrochemical modeling and systematic parameterization for a lithium-ion battery cell[J].Journal of Power Sources,2010,195(15):5071-5080.
[74]Bazant Martin Z.Theory of chemical kinetics and charge transferbased on nonequilibrium thermodynamics[J].Accounts of Chemical Research,2013,46(5):1144-1160.
[75]Jamnik J,Maier J.Charge transport and chemical diffusion involving boundaries[J].Solid State Ionics,1997,94(1-4):189-198.
[76]Jamnik J,Kalnin J R,Kotomin E A,et al.Generalised Maxwell-Garnett equation:Application to electrical and chemical transport[J].Physical Chemistry Chemical Physics,2006,8(11):1310-1314.
[77]Bisquert J.Analysis of the kinetics of ion intercalation:Ion trapping approach to solid-state relaxation processes[J].Electrochimica Acta,2002,47(15):2435-2449.
[78]Jamnik J,Maier J,Pejovnik S.A powerful electrical network model for the impedance of mixed conductors[J].Electrochimica Acta,1999,44(24):4139-4145.
