Microbial Characterization of Denitrifying Sulfide Removal Sludge Using High-Throughput Amplicon Sequencing Method
2015-06-22MaWenjuanLiuChunshuangZhaoDongfengGuoYadongWangAijieJiaKuili
Ma Wenjuan; Liu Chunshuang; Zhao Dongfeng; Guo Yadong; Wang Aijie; Jia Kuili
(1. College of Chemical Engineering, China University of Petroleum, Qingdao 266580; 2. State Key Laboratory of Urban Water Resource and Environment, Harbin Institute of Technology 150090)
Microbial Characterization of Denitrifying Sulfide Removal Sludge Using High-Throughput Amplicon Sequencing Method
Ma Wenjuan1; Liu Chunshuang1; Zhao Dongfeng1; Guo Yadong1; Wang Aijie2; Jia Kuili1
(1. College of Chemical Engineering, China University of Petroleum, Qingdao 266580; 2. State Key Laboratory of Urban Water Resource and Environment, Harbin Institute of Technology 150090)
The denitrifying sulfide removal (DSR) process has recently been studied extensively from an engineering perspective. However, the importance of microbial communities of this process was generally underestimated. In this study, the microbial community structure of a lab-scale DSR reactor was characterized in order to provide a comprehensive insight into the key microbial groups in DSR system. Results from high-throughput sequencing analysis revealed that the fraction of autotrophic denitrifiers increased from 2.34 % to 10.93% and 44.51% in the DSR system when the influent NaCl increased from 0 g/L, to 4 g/L and 30 g/L, respectively. On the contrary, the fraction of heterotrophic denitrifiers decreased from 61.74% to 39.57%, and 24.12%, respectively. Azoarcus and Thiobacillus were the main autotrophic denitrifiers, and Thauera was the main hetetrophic denitrifier during the whole process. This study could be useful for better understanding the interaction between autotrophs and heterotrophs in DSR system.
denitrifying sulfide removal; autotrophic denitrifiers; hetetrophic denitrifiers; microbial communities
1 Introduction
At present, wastewater from the oil industry represents a tremendous challenge for treatment before discharge because of its chemical complexity. This effluent usually contains a high concentration of organic compounds, sulfides and ammonia. Generally some salts like NaCl are also frequently found. Nitrogen compounds contribute mainly to eutrophication of water bodies, besides the risks associated with ammonia toxicity and bad odors. Sulfides are also an environmental problem due to their toxicity, odor and corrosive properties in acidic environments. High salinity may lead to equipment corrosion and other environmental problems.
Denitrifying sulfide removal (DSR) process involves simultaneous removal of nitrogen (NO3-→N2), sulfide (S2-→So) and carbon (acetate→CO2). The DSR process is an attractive option for treating sulfur and carbon compounds-rich wastewater due to its simple processing system, a lower sludge production and the easy recovery of S0[1-3]. Since then Reyes-Avila and Razo-Flores Gomes had achieved simultaneous removal of nitrogen, sulfides, and carbon species using symbiotic heterotrophs and autotrophs in the same CSTR in 2004[4]. Chen, et al. cultivated biogranules in an expanded granular sludge bed (EGSB) reactor and simultaneously removed sulfides, nitrate, and organic carbon species at a high loading rate of 6.0 kg/m3d for sulfides, 3.11 kg/m3d for nitrate, and 3.27 kg/m3d for acetate in 2008. Liu, et al.[5]further demonstrated that the simultaneous removal of nitrate, sulfide and acetate also could be achieved by DSR process at a NaCl concentration of 2—35 g/L. To date, major changes to the design and operation of DSR based systems have been done predominantly from an engineering perspective[6-9], notwithstanding greatly underestimating the importance of microbial communities as an integral component of these systems[10-12]. Therefore, many essential aspects regarding the ecology and dynamics of microbial communities within these systems, necessary for a rational improvement of their operation, remain unresolved.
The high-throughput sequencing technique developed several years ago has exhibited an overwhelming supe-riority on profiling complex bacteria communities for its unprecedented sequencing depth[13]. It can give detailed information on microbial community structure qualitatively and quantitatively, especially in analyzing the subdominant groups at a relative abundance of 0.01—0.1%[14]. So far, this technique has been widely used in investigating microbial diversity and abundance in various samples[15-17]. However, to the best of our knowledge, there have been few papers describing the microbial community structure of the DSR system using the high-throughput sequencing techniques.
In this paper, a DSR process was established and the succession of microbial communities was further investigated using the Illumina MiSeq platform. The objective of this study was to characterize the key microbial groups under different operating conditions.
2 Materials and Methods
2.1 Reactor and synthetic wastewater
The plexiglas EGSB reactor, 50 mm in diameter and 80 cm in height, had a working volume of 1.57 L (Figure 1). The temperature in the reactor was maintained at 30±1 ℃. A peristaltic pump introduced the influent into the reactor at the column bottom. A gas-scrubbing device collected the H2S gas generated at the column overhead. The reflux ratio was fixed at 6:1.

Figure 1 The schematic of experimental setup1—Feed tank; 2—Influent pump; 3—EGSB reactor; 4—Thermostat; 5—Recycle pump; 6—Gas sampler; 7—Wet gas meter; 8—Effluent waste metering tank. A—Water pipeline; B—Gas pipeline.
Seed sludge was collected from the anaerobic sludge thickener at the Nibuwan Wastewater Treatment Plant, Qingdao, China. The seed sludge was screened using a 0.2-mm Tyler-mesh to remove most solids. Synthetic wastewater consisting of 200 mg/L of S2-, 87.5 mg N/L of NO3-, 77.5 mgC/L of acetate and 1500 mg/L of NaHCO3was initially used. The influent pH value was maintained at 7.5.
2.2 Microbial population
Biomass samples from the EGSB reactor were collected and immediately stored at -80 ℃ for subsequent experiments. Total genomic DNA was extracted in duplicate from each sample using the PowerSoil DNA isolation kit (MoBio, Carlsbad, CA) as described in the manufacturer’s instructions, and the extract was subsequently pooled to reduce sample variability. Following extraction, the quality of the DNA was examined by the 1% (w/v) agarose gel electrophoresis and concentration technique measured with a UV-Vis spectrophotometer (NanoDrop 2000, USA). The V3-V4 region of the 16S rRNA gene was amplified using bacterial primers 338F (5’-ACT CCT ACG GGA GGC AGC AG-3’) and 806R (5’-GGA CTA CHV GGG TWT CTA AT-3’)[18], with the reverse primer containing a 6 bp barcode used to tag each sample. PCR amplifications were carried out in triplicate for each sample using 20 mL of reaction mixtures containing 4 mL of 5×PCR buffer, 10 ng of template DNA, 0.2 mM of each primer, 0.25 mM of each dNTP, and 1 U FastPfu polymerase (TransGen, China). The PCR conditions involved an initial denaturation step at 95 ℃ for 2 min, followed by 30 cycles of 95 ℃ for 30 s, 55 ℃ for 30 s, and 72 ℃ for 30 s and ended with an extension step at 72 ℃ for 5 min in a GeneAmp 9700 thermocycler (ABI, USA). The triplicate amplicos were pooled together, electrophoresed on a 2% (w/v) agarose gel, and recovered using an AxyPrep DNA Gel Extraction kit (AXYGEN, China).
The purified amplicon was quantified by QuantiFluor-ST Fluorometer (Promega, USA), and then a composite sequencing library was constructed by combining equimolar ratios of amplicons from all samples. The resulting library was sent for paired-end sequencing (2×250 bp) on an Illumina MiSeq platform at Majorbio Bio-Pharm Technology Co., Ltd. (Shanghai, China).Low quality reads (ambiguous nucleotides with a quality value of <20) were removed from the raw sequence data as described in the literature[19]. The paired-end reads from each sample were verlapped to assembly V3—V4 tags of 16S rRNA gene using SeqPrep (https:// github. com/jstjohn/SeqPrep), and then UCHIME was used to remove chimera sequences from the tags (http://drive5. com/ usearch/manual/uchime_algo.html). To facilitate the comparison between different samples, the number of sequences was normalized to the same sequencing depth of 43,900 by the MOTHUR program[20]. The effective sequences were clustered into operational taxonomic units (OTUs) at 97% sequence identity using UPARSE embedded in Qiime[21], and a representative sequence was then picked for each OTU by selecting the most abundant sequence in that OTU. These representative sequences were assigned to taxonomic classifications by RDP Classifier with a confidence threshold of 70%[22]. Additionally, the alpha diversity including rarefaction curves, Shannon diversity index and diversity coverage were calculated in MOTHUR for each sample. Cluster analysis based on the Bray-Curtis similarity index was also carried out using PAST (http://folk.uio.no/ohammer/past/). Raw sequence data of this study have been deposited to the NCBI Sequence Read Archive with the accession No. SRS832283, No. SRS832287, No. SRS833526, and No. SRS833528, respectively.
2.3 Chemical analysis
The concentrations of nitrate, nitrite, sulfate, and thiosulfate in the collected liquor samples following 0.45 mm filtration were measured by ion chromatography (ICS-3000; Dionex, USA). The sample separation and elution were performed using an IonOac AG4AAS4A-SC 4 mm analytical column with carbonate/bicarbonate eluent (1.8 mmol/L of Na2CO3and 1.7mmol/L of NaHCO3at a rate of 1 cm3/min) and sulfuric acid regeneration (25 mmol/L of H2SO4at a rate of 5 cm3/min). The sul fide concentration was determined using the methylene blue method[23]. The term ‘sulfide’ in this study includes all species in the liquid sample (H2S(aq), HS-and S2-). The acetate concentration was determined by gas chromatography (type 6890; Agilent, USA). A pH meter (pHS-25; Shanghai, China) determined the pH value of the liquid samples. The amounts of suspended solids and VSS were measured according to the standard methods referred to in the literature[24].
3 Results and Discussion
3.1 Reactor performance
The initial synthetic wastewater was introduced into EGSB reactor at an initial loading rates of: 0.4 kgS/(m3·d) for sulfide, 0.175 kgN/(m3·d) for nitrate, 0.15 kgC/(m3·d) for acetate in the absence of NaCl for 20 d (Table 1). Then the influent NaCl concentration was increased gradually from 4 g/L to 30 g/L and the influent acetate loading rate was increased to 0.45 kgC/(m3·d). High efficiency for removal of sulfides was maintained during the whole process (100%). Nitrate was also completely removed from the DSR system. Additionally, the efficiency for removal of acetate showed a slight decrease, but was still maintained at 85.2% after the acetate loading rate increased to 0.45 kgC/(m3·d). Straw-yellow, granular and filamentous insoluble elemental sulfur (S0) solids were present in the reactor. Most sulfide species removed were converted to S0(100%). The above-mentioned phenomenon indicated that a high DSR performance was achieved regardless ofthe NaCl concentration.

Table 1 Operational data and reactor performance of denitrifying sulfide removal reactor
3.2 Diversity of microbial community identified by high-throughput sequencing
The parameters related to the alpha diversity of microbial community for each sample at a distance cutoff level of 0.03 are shown in Table 2. The species richness for bacteria in the reactor varied significantly during the 112-day operation, which was revealed by OTUs and Chao 1. As shown in Figure 2, the rarefaction curves of the four samples at 0.03 distance suggested that the sequencing depths for all samples were well enough to cover the whole diversity. This was confirmed by the coverage values of the four samples (99.90%, 99.31%, 99.82% and 99.50%, respectively, as shown in Table 2), indicating that almost all of OTUs in the reactor were detected in this study. The results of Shannon diversity index in Table 2 also demonstrated that the bacterial diversity in the reactor sunk slightly from 3.91 to 2.64. In addition, as shown in Figure 2, the cluster analysis indicated that a great change in microbial community composition occurred during the whole operation period of DSR reactor.

Figure 2 Rarefaction curves based on the sequencing of bacterial communities1, 2, 3 and 4 were collected at days 0, 20, 62 and 112, respectively. The OTUs were defined by 0.03 distance.
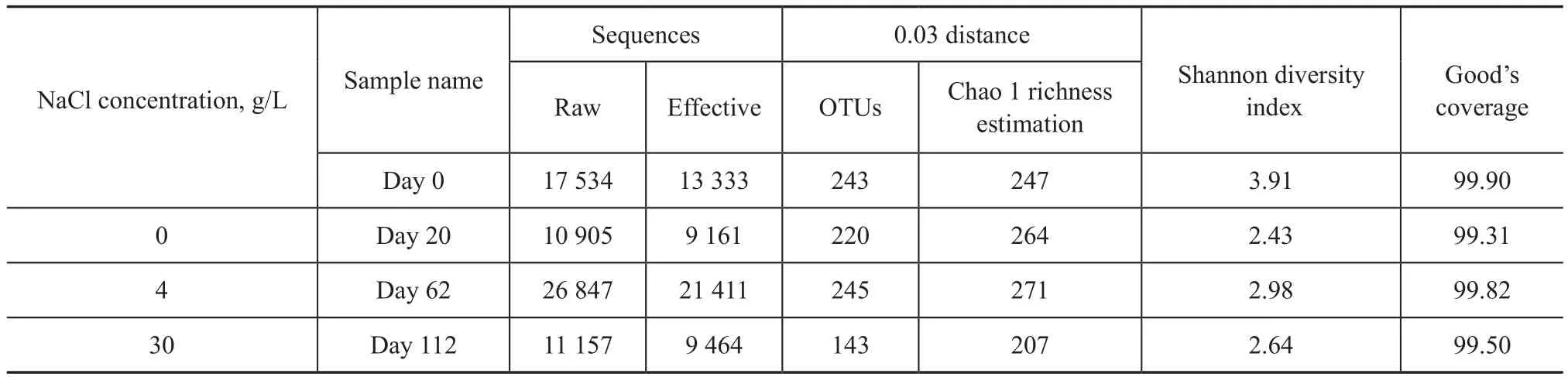
Table 2 Richness and diversity of the four samples based on 0.03 distance
3.3 Microbial community composition and dynamics
The phylogenetic classification of effective bacterial sequences from the four samples at two different taxonomic levels (phylum and genus, respectively) is summarized in Figure 3, demonstrating distinct bacterial community compositions. It is found from the phylum assignment result (Figure 3a) that bacterial diversity in the reactor shifted greatly during the 112 days of operation. In total, 9 known bacterial phyla were detected in the seeding sludge (day 0), which mainly included Proteobacteria, Bacteroidetes, Acidobacteria, Actinobacteria, Nitrospirae, and Chloroflexi, which accounted for 69.5%, 14.7%, 4.51%, 2.75%, 2.60% and 2.11%, respectively. Most of bacterial phyla were reduced steeply and would eventually vanish during the 112-day operation, except for Proteobacteria and Bacteroidetes. The abundance of Proteobacteria significantly decreased and remained stable at 54%, while the abundance of Bacteroidetes underwent a slight decrease to 2%. Besides, there was a remarkable increase of Firmicutes from 0.4% to 38.9% over 62 days, and then it decreased slightly to 6.4% on the 112thday. In all, only three phyla, including Proteobacteria, Bacteroidetes and Firmicutes, were selected to establish a stable foothold in the community, although high diversity was identified in the inoculum. Such selection was probably driven by the availability of substrate in the influent.
Analyzing at the genus level can bring us around to a deeper understanding of the community function in the microbial ecosystem (Figure 3b). Many sequences could not be classified at the genus level (14.69%, 6.77%, 5.15% and 8.36% for day 0, day 20, day 62 and day 112, respectively) which indicated that many taxa remainedunknown in the reactor. However, it is worth mentioning that a majority of these unknown taxa belonged to phylum Rhodobacteraceae, characterized as typical heterotrophic denitrification bacteria in activated sludge. A wide range of bacteria genera were identified as dominant in the inoculum, such as Xanthomonadales (9.08%), Piscinibacter (7.10%), C1-B045 (6.70%) and Denitratisoma (5.39%), whereas almost all of them were eliminated after the startup of the reactor.
The genera Thauera, Anaerobacillus, Thermomonas and Aquimonas, which were reported commonly as hetetrophic denitrifiers, changed diversely with the influent NaCl concentration increasing from 0 g/L to 30 g/L. The dominant genera Thauera and Thermomonas were decreased from 39.245% and 22.49% to 15.18% and 1.01%, respectively, with the influent NaCl concentration increasing from 0 g/L to 35 g/L. There was a remarkable increase of Anaerobacillus from 0% to 14.86% with the influent NaCl concentration increasing from 0 g/L to 4 g/L, then decreased to 1.06% at an influent NaCl concentration of 35 g/L. In contrast, the abundance of Aquimonas significantly increased from 0 to 6.85% with the influent NaCl concentration increasing from 0 g/L to 35 g/L. Surprisingly, the OTUs that most closely reacted on Azoarcus and Thiobacillus, which were reported commonly as autotrophic denitrifiers, increased prominently when NaCl concentration increased from 0 g/L to 35g/L.
In all, the fraction of heterotrophic denitrifers decreased from 61.74% to 39.57% and 24.12% with the influent NaCl increasing from 0 g/L to 4 g/L and 30 g/L, respectively. On the contrary, the fraction of autotrophic denitrifiers increased from 2.34 % to 10.93% and 44.51%, respectively. Nonetheless, stable and balanced nitrogen and sulfur removal could be obtained in all circumstances. The reason may be that the addition of NaCl caused the decrease in the activity of heterotrophic denitrifiers, while it might have little effect on autotrophic denitrifiers. Some species belonging to Thiobacillus could better grow withthe 0.4—1.0 mol/L NaCl solution[25]. With an increasing NaCl concentration, a certain heterotrophic denitrifier which could tolerate high NaCl concentration was selected, such as some species belonging to Aquimonas[26]. And some species belonging to Azoarcus, which was dominant when NaCl concentration was 30 g/L, could also grow by facultative denitrification[27]. Therefore, high DSR performance could also be maintained regardless of the concentration of NaCl in DSR system.

Figure 3 Taxonomic classification of the bacterial communities at (a) phylum and (b) genus levels. Phylum and genus making up less than 0.5% of total composition in the sample was classified as “others”
4 Conclusions
In summary, this study gives a detailed insight into the bacterial community composition of the DSR process. The fraction of autotrophic denitrifiers increased from 2.34 % to 10.93% and 44.51% in the DSR system, when the influent NaCl concentration increased from 0 mg/L, to 4 g/L and 30 g/L, respectively. On the contrary, the fraction of heterotrophic denitrifiers decreased from 61.74% to 39.57% and 24.12%, respectively. High DSR performance could also be maintained regardless of the concentration of NaCl solution. Azoarcus and Thiobacillus were the main autotrophic denitrifiers, and Thauera was the main hetetrophic denitrifier identified during the whole process.
Acknowledgement:The research was supported by the National Natural Science Foundation of China under Grant No. 21307160, the Natural Science Foundation of Shandong Province under Grant No. ZR2013EEQ030 and the Fundamental Research Funds for the Central Universities under Grant No. R1404005A.
[1] Wang A J, Du D Z, Ren N Q, et al. An innovative process of simultaneous desulfurization and denitrification by Thiobacillus denitrifier[J]. J Environ Sci Health A, 2005, 40(10): 1939-1949
[2] Wang A J, Liu C S, Han H J, et al. Modeling denitrifying sulfide removal process using artificial neural networks[J]. J Hazard Mater, 2009, 168(2/3): 1274-1279
[3] Chen C, Ren N Q, Wang A J, et al. Microbial community of granules in EGSB reactor for simultaneous biological removal of sulfate, nitrate, and lactate[J]. Appl Microbiol Biotechnol, 2008, 79(6): 1071-1077
[4] Reyes-Avila J, Razo-Flores G. Simultaneous biological removal of nitrogen, carbon and sulfur by denitrification[J]. Water Res, 2004, 38(14/15): 3313-3321
[5] Liu Chunshuang; Zhao Dongfeng; Zhang Yunbo. Effects of temperature, acetate and nitrate on methane generation from petroleum hydrocarbons[J]. China Petroleum Processing and Petrochemical Technology, 2014, 16(4): 24-31
[6] Chen C, Ren N Q, Wang A J, et al. Simultaneous biological removal of sulfur, nitrogen and carbon using EGSB reactor[J]. Appl Microbiol Biotechnol, 2008, 78(6): 1057-1063
[7] Zhou X, Chen C, Wang A J, et al. Biosorption of Cu(II) by powdered anaerobic granular sludge from aqueous medium[J]. Water Sci Technol, 2013, 68(1): 91-98
[8] Xu X, Chen C, Wang A J, et al. Simultaneous removal of sulfide, nitrate and acetate under denitrifying sulfide removal condition: Modeling and experimental validation[J]. J Hazard Mater, 2014, 264: 16-24
[9] Chen C, Liu L, Lee D J, et al. Integrated simultaneous desulfurization and denitrification (ISDD) process at various COD/sulfate ratios[J]. Bioresour Technol, 2014, 155: 161-169
[10] Xu X, Chen C, Wang A, et al. Bioreactor performance and functional gene analysis of microbial community in a limited-oxygen fed bioreactor for co-reduction of sulfate and nitrate with high organic input[J]. J Hazard Mater, 2014, 278: 250-257
[11] Yu H, Chen C, Ma J, et al. Geochip-based analysis of the microbial community functional structures in simultaneous desulfurization and denitrification process[J]. J Environ Sci-China, 2014, 26(7): 1375-1382
[12] Yu H, Wang A, Chen C. Structure and dynamic of microbial community in the denitrifying sulfide removal process[J]. J Environ Sci-China, 2013, 34 (3): 1190-1195
[13] Glenn T C. Field guide to next-generation DNA sequencers[J]. Mol Ecol Resour, 2011, 11(5): 759-769
[14] Guo F, Zhang T. Profiling bulking and foaming bacteria in activated sludge by high throughput sequencing[J]. Water Res, 2012, 46(8): 2772-2782
[15] Lu L, Xing D, Ren N. Pyrosequencing reveals highly diverse microbial communities in microbial electrolysis cells involved in enhanced H2production from waste activated sludge[J].Water Res, 2012, 46(7): 2425-2434
[16] Ye L, Zhang T. Bacterial communities in different sections of a municipal wastewater treatment plant revealed by 16SrDNA 454 pyrosequencing[J]. Appl Microbiol Biotechnol, 2013, 97(6): 2681-2690
[17] Filbert E M, Agrawal S, Karst S M, et al. Low temperature partial nitritation/anammox in a moving bed biofilm reactor treating low strength wastewater[J]. Environ Sci Technol, 2014, 48(15): 8784-8792
[18] Claesson M J, Wang Q, Sullivan O O, et al. Comparison of two next-generation sequencing technologies for resolving highly complex microbiota composition using tandem variable 16SrRNA gene regions[J]. Nucleic Acids Res, 2010, 38(2): 1-13
[19] Caporaso J G, Lauber C L, Walters W A, et al. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample[J]. Proc Natl Acad Sci USA, 2011, 108(1): 4516-4522
[20] Schloss P D, Westcott S L, Ryabin T, et al. Introducing mothur: open-source, platform-independent, communitysupported software for describing and comparing microbial communities[J]. Appl Environ Microbiol, 2009, 75(23): 7537-7541
[21] Caporaso J G, Kuczynski J, Stombaugh J, et al. QIIME allows analysis of high-throughput community sequencing data[J]. Nat Methods, 2010, 7(5): 335-336
[22] Wang Q, Garrity G M, Tiedje J M, et al. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy[J]. Appl Environ Microbiol, 2007, 73(16): 5261-5267
[23] APHA. Standard Methods for the Examination of Water and Wastewater[M], 21sted. American Water Works Association and Water Environment Federation, Washington, DC, USA. 2005
[24] Tang C J, Zheng P, Mahmood Q, et al. Start-up and inhibition analysis of the anammox process seeded with anaerobic granular sludge[J]. J Ind Microbiol Biotechnol, 2009, 36(8): 1093-1100
[25] Paulina K. The effect of some salts on Thiobacillus thioparus[J]. Can J Microbiol, 1969, 15(3): 314-318
[26] Krieger B, Schwermer C U. Diversity of nitrate-reducing and denitrifying bacteria in a marine aquaculture biofilter[C]. Proceedings of the 11th International Symposium on Microbial Ecology (ISME-11). International Society for Microbial Ecology, 2006
[27] Lee D J, Wong B T, Adav S S. Azoarcus taiwanensis sp. nov., a denitrifying species isolated from a hot spring[J]. Appl Microbiol and Biotechnol, 2014, 98(3): 1301-1307
Iron-based Fischer-Tropsch Synthesis Catalyst Achieves an Alcohol Selectivity Exceeding 60%
The key project “Nano-scale catalyst and related process research and development activity for high-efficiency synthesis of mixed lower alcohols from syngas” undertaken by the Shanghai Senior Research Institute of CAS has passed the acceptance tests by the expert group organized by the Shanghai Science and Technology Commission. It is said that this new-generation catalyst has displayed its excellent product selectivity including an over 60% selectivity of total alcohols, among which the selectivity of C2+ higher alcohols exceeds more than 90%.
The research team through designing a nano-structure of the catalyst has significantly enhance its reaction performance while concurrently inhibiting the formation of methane and CO2(with total CH4+CO2selectivity being less than 5%) to make breakthroughs in terms of a continuous and smooth operation exceeding 8 000 hours. Consequently, the Fe-based F-T synthesis catalyst operating at low temperature with high carbon efficiency has been successfully prepared. Meanwhile, in order to directly synthesize olefins from syngas (FTO), the research team by means of enhancing the interaction of components has made success in selectively suppressing the methane formation and secondary hydrogenation of olefins, resulting in low methane selectivity (around 5%) and high olefin selectivity (>80%) under mild conditions to lay a solid foundation for the development of FTO catalysts with high selectivity at low temperature.
date: 2015-03-03; Accepted date: 2015-08-20.
Liu Chunshuang, Telephone: +86-532-86982875; Fax: +86-532-86982875; E-mail: liuchunshuang 723@126.com.
杂志排行

中国炼油与石油化工的其它文章
- Computational Fluid Dynamics Simulation of Liquid-Phase FCC Diesel Hydrotreating in Tubular Reactor
- Quantitative Analysis Using Fourier Transform Ion Cyclotron Resonance Mass Spectrometry and Correlation between Mass Spectrometry Data and Sulfur Content of Crude Oils
- Hydrothermal Liquefaction of Wheat Straw in Sub-critical Water/Ethanol with Ionic Liquid for Bio-oil Production
- Promotional Effect of CoO(OH) on Selective Hydrogenation of Maleic Anhydride to γ-Butyrolactone over Supported Ruthenium Catalyst
- Synthesis and Separation Performance of Y-type Zeolite Membranes by Pre-Seeding Using Electrophoresis Deposition Method
- Design and Control of Self-Heat Recuperative Distillation Process for Separation of Close-Boiling Mixtures: n-Butanol and iso-Butanol