协同场作用下钒离子在质子交换膜中的传质过程
2015-04-01张友王树博齐亮姚克俭
张友,王树博,齐亮,姚克俭
(1浙江工业大学化工学院绿色化学合成技术国家重点实验室培育基地,浙江 杭州 310032;2清华大学核能与新能源技术研究院,北京 100084)
引言
全钒液流电池储能技术是一种新型的高效电化学储能技术,与其他储能电池相比,它具有如下优势:能量转换效率高、蓄电容量大、系统设计灵活、可靠性高、可深度充放电等一系列优点,被认为是最有前景的储能技术[1-7]。
全钒液流电池主要由电极、电解液、质子交换膜和集流体等几个关键部分构成,随着近年来关键材料领域不断取得技术突破[8-14],目前的研究热点逐渐集中到了电池结构优化设计及对电池内部传递过程的研究[15-17]。理想情况下,质子交换膜既能为正负电解液提供质子传递的通道,又能阻隔正负电极电解液的相互混合[18]。事实上,在电池运行过程中,电解质跨膜渗透问题无法完全避免,正负电解质的混合导致的电池自放电现象,是引起的电池容量损失进而导致电池性能恶化的重要原因[19-20],所以无论是在建立电池模型还是研究电池极化以及电池性能寿命等课题上,电池隔膜中的离子传递问题都无法回避。关于电解质中钒离子透膜传质问题,前人已经有过相关研究[21-24]。但仅限于考虑质子交换膜中单纯的浓差扩散过程,往往通过直接测得表观扩散系数来研究钒离子跨膜传质过程,建立的相关模型也主要是基于 Fick定律离子扩散模型推导得到,这并不能完全与电池在运行中的实际状况符合,难以有实际应用价值。因为,钒离子在质子交换膜中的传质过程,是处于电场环境之下,由浓差推动力和电场力协同作用的结果,所以除了由于膜两侧浓度不同而造成的浓差扩散外,还应该考虑钒离子在场下的电迁移作用。研究这种多物理场耦合作用下离子传质过程对于提高电池性能、优化电池结构、提高电池容量和充放电特性以及循环寿命,具有非常重要的意义,也为扫清全钒液流电池商业化应用的技术障碍提供了有益的理论基础。本文正是从协同场这一角度,通过设计外加电场作用下钒离子跨膜渗透实验,系统研究了不同工况下钒离子在质子交换膜中的传质过程,并测得了四价钒离子的表观电迁移率。
1 数学模型
对于钒离子在Nafion膜中的传质过程,如果只考虑单纯的浓差扩散,由Fick定律可知扩散传质速率j(d)
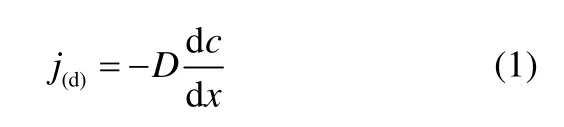
在假设膜左侧浓度c1不变的情况下,通过积分处理可得扩散系数D
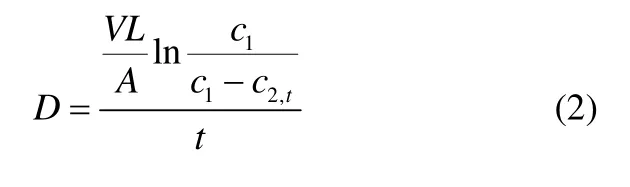
在电场作用下,钒离子的电迁移传质速率为j(e),其数学形式为
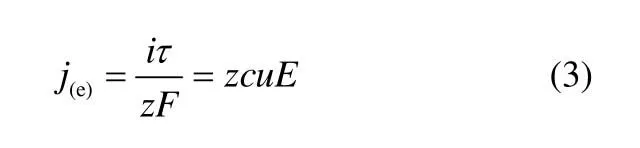
综合膜中考虑扩散和电迁移这两种传质形式,总的传质速率公式可以表示为
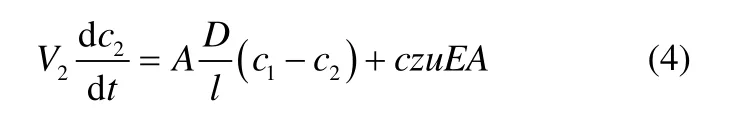
式中,c1和c2是膜两侧的浓度;c为质子交换膜中钒离子的平均浓度。
因为膜的厚度非常薄,可以认为膜中从高浓度一侧到低浓度一侧的浓度变化是近似线性的,则式(4)中

模型中,若不考虑由于少量水跨膜传质导致的电解液体积变化,根据钒离子的物质守恒,有

通过以上公式,可以求得在质子交换膜中钒离子的电迁移率
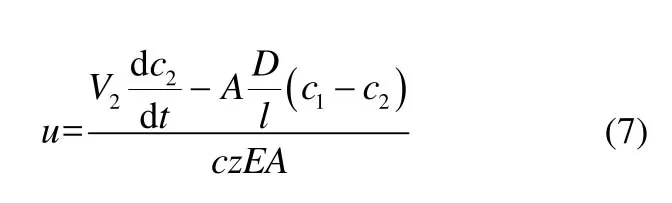
为了衡量电场作用对钒离子跨膜传质通量的影响,引入电场因子St的概念,其定义为电场存在下的传质通量与单纯浓差扩散传质通量之比
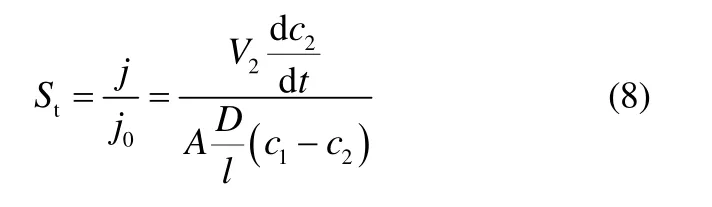
2 实 验
2.1 仪器及药品
仪器:高精度电池性能测试系统(深圳市新威尔电子有限公司)、紫外可见分光光度计(岛津UV-1750)、自动电位滴定仪(上海雷磁)、多功能磁力搅拌器、恒温水浴锅、蠕动泵、石墨电极、电热鼓风干燥箱、自制电解槽、Nafion117膜。
药品:硫酸氧钒(VOSO4·3H2O)(沈阳市海中天精细化工厂)、硫酸(北京化工厂)、无水硫酸镁(国药集团化学试剂有限公司)。
2.2 实验方案
膜的预处理:在每一组实验前将Nafion117质子交换膜在质量分数为 5%的双氧水中 80℃煮沸1 h,用去离子水冲洗3次后,放入去离子水中80℃下煮沸1 h,再放入0.5 mol·L-1的 H2SO4溶液中80℃煮沸1 h,最后再在去离子水中80℃煮沸1 h,结束时用去离子水浸泡备用。
标准曲线的绘制:配制一系列标准浓度的VOSO4的硫酸溶液,应用相应浓度的 MgSO4溶液作为参比溶液,用紫外分光光度计测出其在波长为762 nm处的吸光度,拟合出标准曲线。
按照图1所示的装置进行渗透性实验:电解槽左侧为 VOSO4与 H2SO4的混合溶液,右侧为相应离子强度的MgSO4与H2SO4的混合溶液。在不同的实验工况下,每隔一段时间,用紫外分光光度计检测右侧电解液(透过侧)中VO2+的浓度,比较VO2+在不同实验工况下的渗透性,并计算其表观电迁移率(离子淌度)。
实验中的参数见表1。
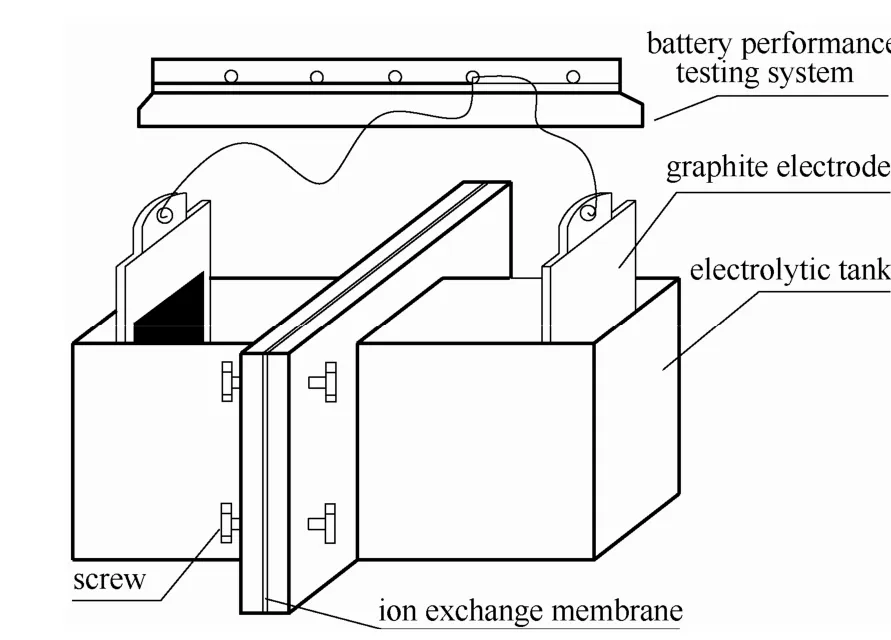
图1 钒电池膜渗透实验装置Fig.1 Schematic diagram of membrane osmotic apparatus of VFB
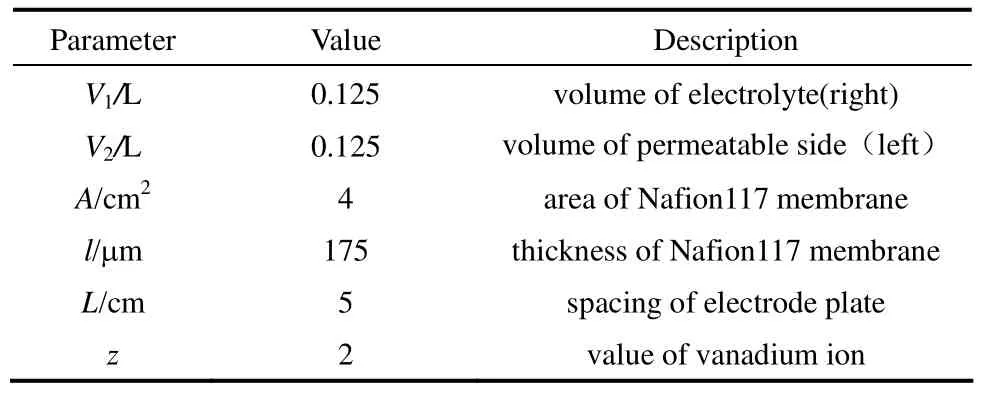
表1 实验中的有关参数Table 1 Parameters in experiment
3 结果与讨论
3.1 无电场作用下膜中传质过程
按照图1的实验装置进行渗透性实验,在无外在电场加入时,测试了不同温度、不同浓度、不同对流状态等操作工况下,VO2+的跨膜渗透特性,实验结果如图2所示。从图中可以看出,对于无电场作用下的渗透传质,各不同操作工况下,透过侧的浓度均随着时间的增加而增加。在时间为32 h时,电解液温度为 45℃且有强制对流(250 r·min-1)存在的情况下,透过侧VO2+浓度甚至达到了0.012 mol·L-1,升高温度和增加溶液中电解液的湍动,都会加剧电解液的渗透。这是因为温度增加,VO2+运动更加剧烈,扩散加快。此外,温度升高,会导致膜体积膨胀,使得Nafion膜致密度下降,从而促进了离子的传质过程。当电解液被强制湍动时,电解液与膜接触面的滞流层厚度减小,滞流层内的浓度梯度降低,这也加剧了VO2+的跨膜渗透过程。
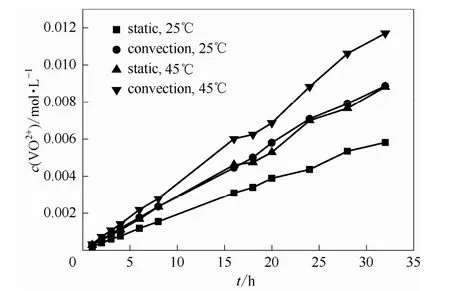
图2 无电场作用时渗透侧VO2+的浓度随时间的变化关系Fig.2 Relationship between VO2+concentration with time inosmotic side without electric field
通过实验测定的不同时刻透过侧的 VO2+的浓度,能够关联出VO2+的扩散系数D,图3为由透过侧浓度求扩散系数D的关联图。表2所示为不同工况下浓差扩散系数D的数值大小。
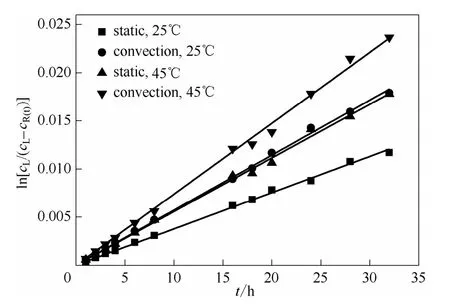
图3 无电场作用时由渗透侧VO2+的浓度关联扩散系数图Fig.3 Diffusion coefficient associated with VO2+concentration in osmotic side without electric field
3.2 电场因素对膜中传质过程的影响
3.2.1 正向电场 全钒液流电池在实际运行过程中,在电场力作用下的电迁移传质过程会对钒离子跨膜渗透产生影响。采用外加恒压电场的方法,可以很好地等效钒离子真实的传质过程。
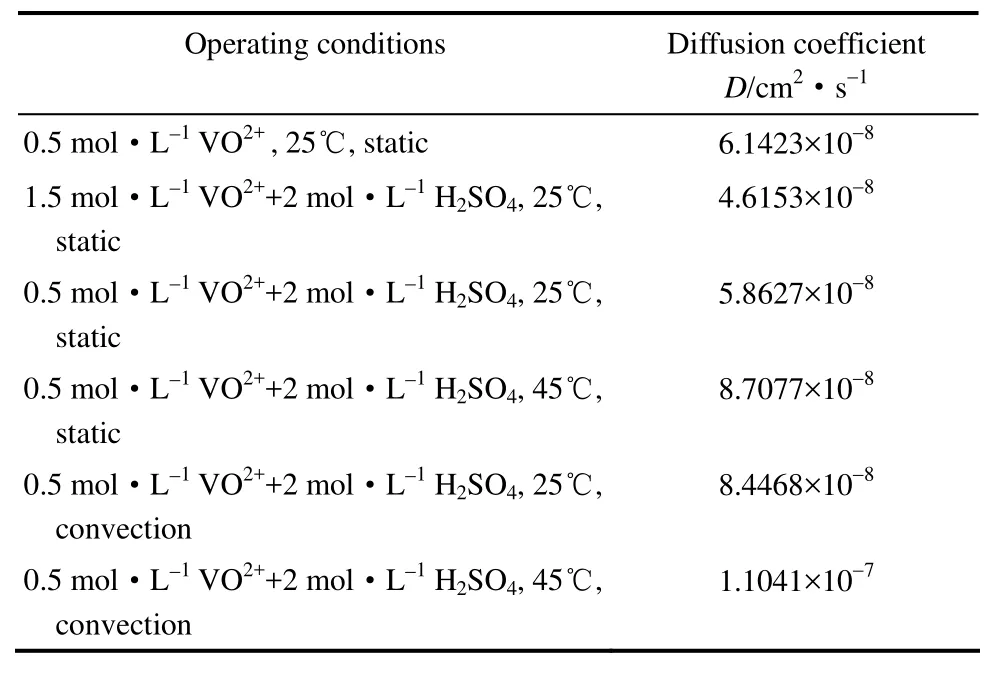
表2 无电场作用时渗透侧VO2+在Nafion117膜中的扩散系数Table 2 Diffusion coefficient of VO2+in Nafion117 membrane without electric field
图 4表示左侧为 0.5 mol·L-1VOSO4与 2 mol·L-1H2SO4右侧为 0.5 mol·L-1MgSO4溶液的体系,在不同外加电压下不同时刻透过侧的 VO2+浓度。图中随着外加电压从0 V增加到3 V,透过侧VO2+的浓度随时间增加的趋势明显,这与理论预期相吻合。
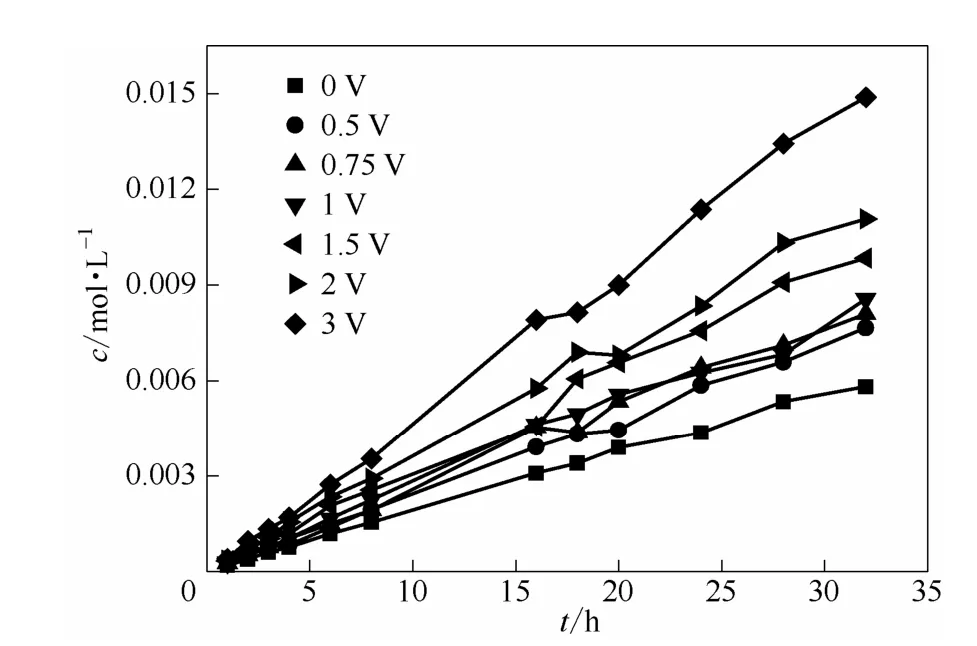
图 4 正向电场下 0.5 mol·L-1VO2++2 mol·L-1H2SO4体系渗透侧VO2+浓度随时间的变化关系Fig.4 Relationship between VO2+concentration with time in osmotic side of 0.5 mol·L-1VO2++2 mol·L-1H2SO4system in positive electric field
图5表示在各不同外加电压下电场因子的随时间的变化趋势。从图中可以发现,外加电压越大,电场因子St越大,电压为0.5 V时,St为1.2左右,当电压增加到3 V时,St可达到2.35~2.41,VO2+发生严重的跨膜渗透传质现象。在不同电压下,电场因子St均随着时间的增加而增大,且随着外电压的加大,St随时间增大的速度越来越快。
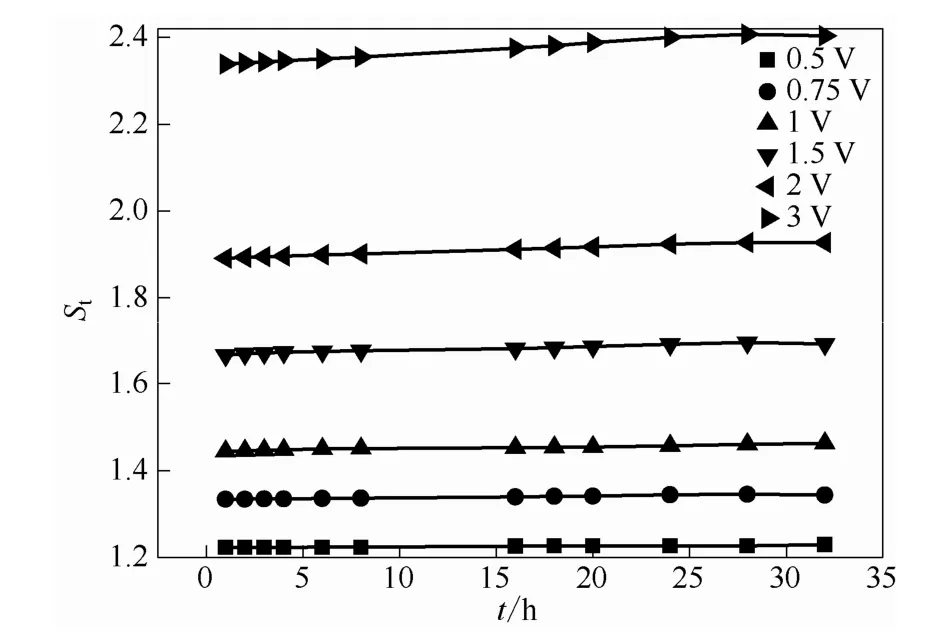
图 5 正向电场下 0.5 mol·L-1VO2++2 mol·L-1H2SO4体系电场因子随时间的变化关系Fig.5 Relationship between electric factor with time of 0.5 mol·L-1VO2++2 mol·L-1H2SO4system in positive electric field
3.2.2 反向电场 当外加电场为反向电场时,如图6所示,电池透过侧的VO2+浓度随着反向电场数值的增大而减小。图 7所示为电场因子在不同反向电压下随时间的变化,可以看出:当反向电压从0.5 V增加到1 V时,St从0.78左右降到0.55左右,这与理论预期相吻合。说明反向电场在抑制离子跨膜渗透防止两极交叉污染方面有一定的应用价值。
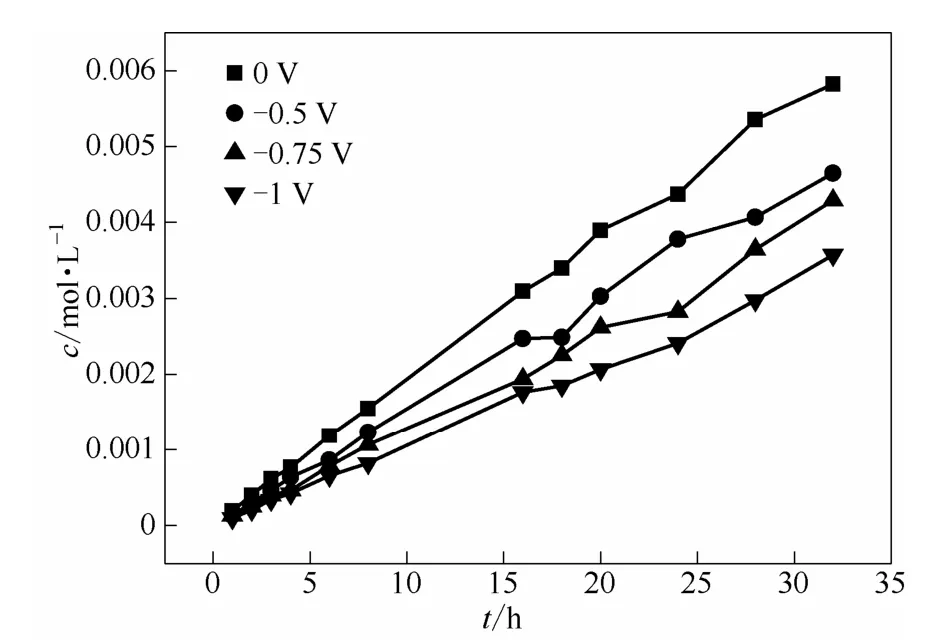
图 6 反向电场下 0.5 mol·L-1VO2++2 mol·L-1H2SO4体系渗透侧VO2+浓度随时间的变化关系Fig.6 Relationship between VO2+concentration with time in osmotic side of 0.5 mol·L-1VO2++2 mol·L-1H2SO4system inreversed electric field
3.3 离子浓度对协同场下膜中传质过程的影响
实验还考察了 VO2+和 H2SO4浓度对协同场下离子在膜中传质过程的影响。
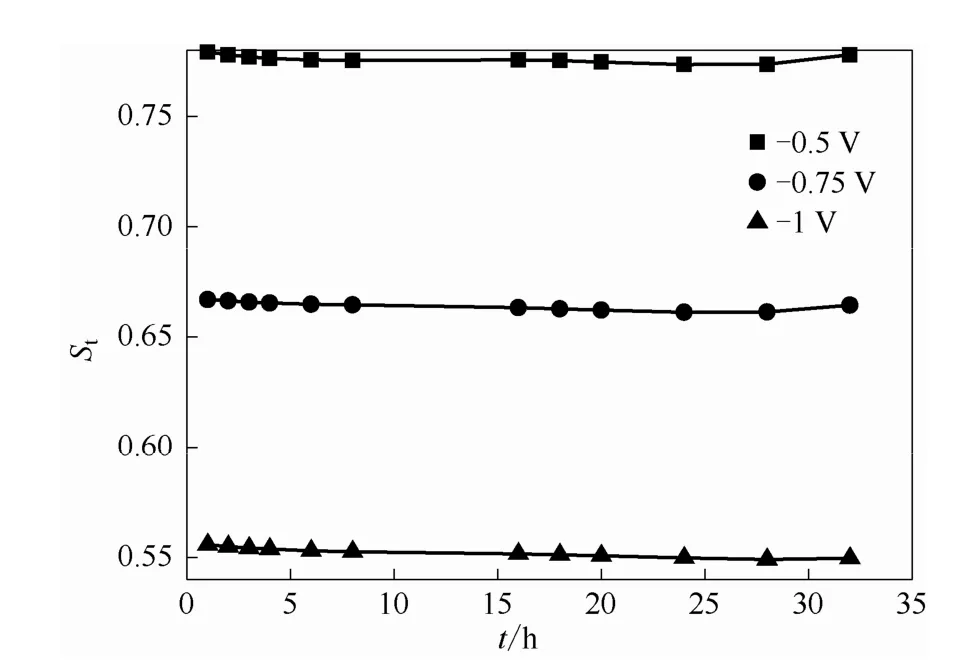
图 7 反向电场下 0.5 mol·L-1VO2++2 mol·L-1H2SO4体系电场因子随时间的变化关系Fig.7 Relationship between electric factor with time of 0.5 mol·L-1VO2++2 mol·L-1H2SO4system in reversed electric field
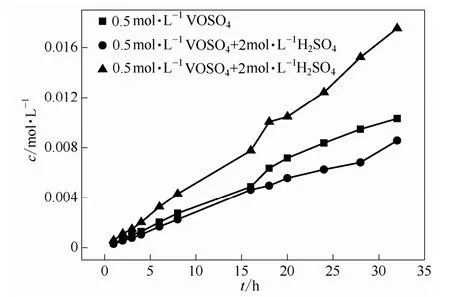
图8 电压为1 V时不同浓度体系渗透侧VO2+的浓度随时间的变化关系Fig.8 Relationship between VO2+concentration with time in osmotic side of different concentration system when voltage is 1 V
如图8所示,当VO2+浓度同样为0.5 mol·L-1时,在透过侧,加入2 mol·L-1H2SO4的实验组比不加 H2SO4的实验组的 VO2+要小,说明 H2SO4的加入有助于抑制电场中VO2+在膜中的传质过程,这是因为电解液中 H+能够比较容易地透过质子交换膜,也就占据了更多的膜内通道,客观上阻碍了钒离子跨膜渗透。
再考察VO2+浓度因素的影响,由于加入侧初始VO2+浓度不同,1.5 mol·L-1VO2++2 mol·L-1H2SO4体系在透过侧的VO2+浓度比0.5 mol·L-1VO2++2 mol·L-1H2SO4体系要大,但是VO2+跨膜传质的程度要轻微得多,这正如图 9所示,电场对于不含H2SO4的体系跨膜传质过程的强化作用最强,在本实验组中,1 V电压的St能够达到1.67~1.71,对于较高浓度的VO2+和H2SO4的体系影响最小,1 V电压的St仅为1.16左右,也即高浓度电解液在真实运行中的电场环境中,有助于降低正负极交叉污染现象。
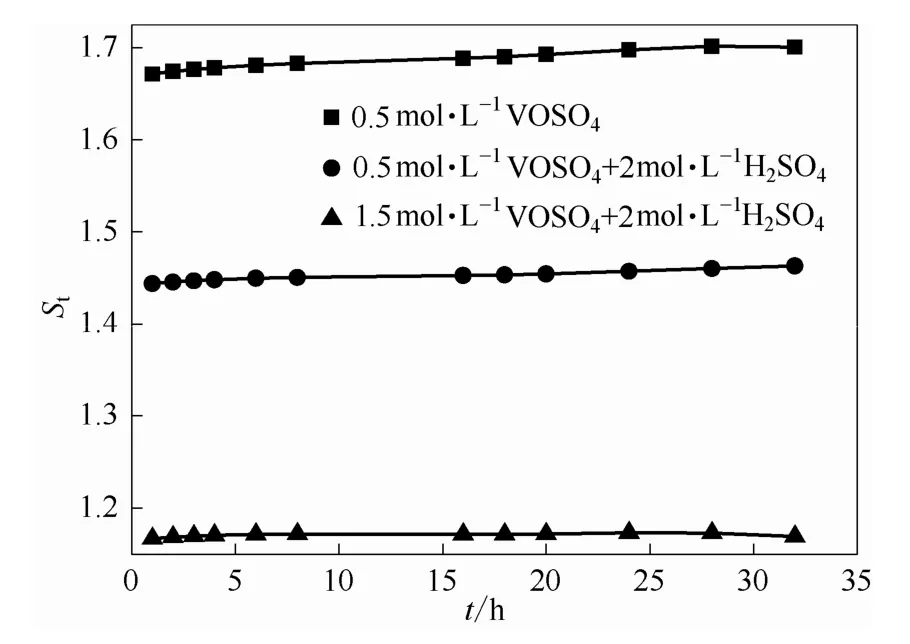
图9 电压为1 V时不同浓度体系电场因子随时间的变化关系Fig.9 Relationship between electric factor of different concentration system when voltage is 1 V
而且有文献报道[25],当H2SO4的浓度增加时,也有利于减小膜电阻,这是因为,当溶液中的 H+浓度较大时,由于 Donnan平衡效应,离子交换膜中的 H+也相应增加,故 H+的迁移速度也会加快,也即膜面电阻减小,膜的电导性增加,这有助于电池性能的提高。
3.4 温度对协同场下膜中传质过程的影响
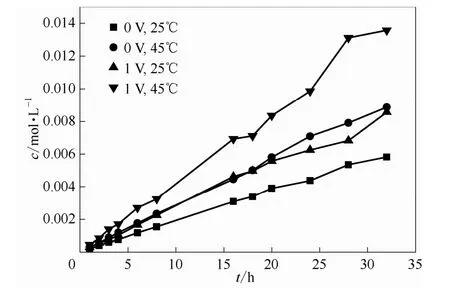
图10 不同温度下0.5 mol·L-1VO2++2 mol·L-1H2SO4体系渗透侧VO2+浓度随时间的变化关系Fig.10 Relationship between VO2+concentration with time in osmotic side of 0.5 mol·L-1VO2++2 mol·L-1H2SO4system in different temperature
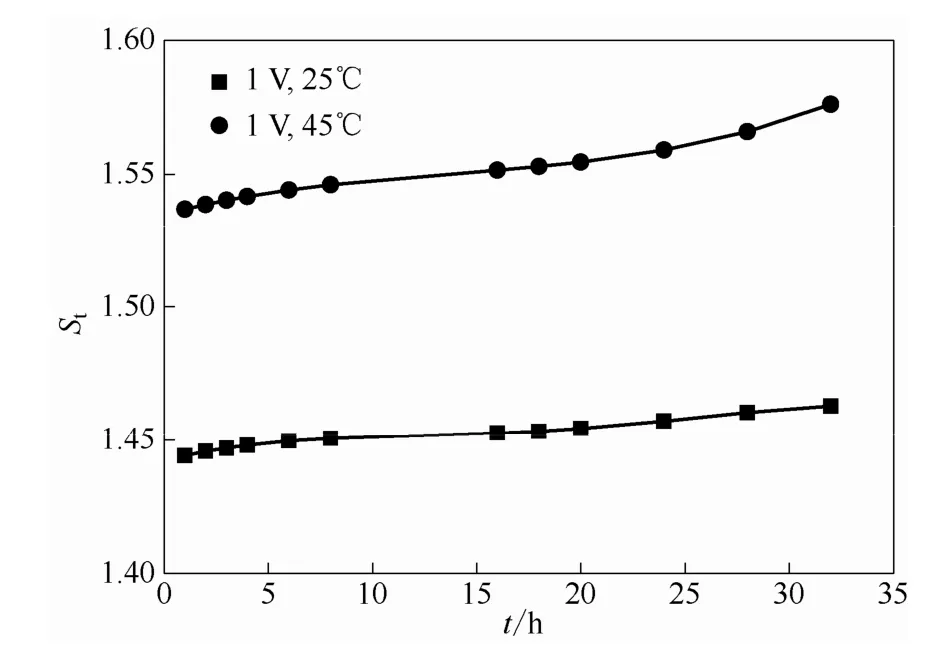
图11 不同温度下0.5 mol·L-1VO2++2 mol·L-1H2SO4体系电场因子随时间的变化关系Fig.11 Relationship between electric factor with time of 0.5 mol·L-1VO2++2 mol·L-1H2SO4system in different temperature
在钒电池运行过程中,温度的影响至关重要,温度升高,离子电迁移率增加,电迁移速度加快,此外温度升高会使离子运动加剧,并且会导致膜的致密性降低,这均会使得钒离子在膜两侧相互渗透的现象严重化;而且在电池真实运行状况中,温度过高,会导致正极的五价钒离子VO2+发生聚集,致使电解液性能下降,这会严重影响钒电池的正常运行。图10显示了静态下电池左侧为0.5 mol·L-1VOSO4与 2 mol·L-1H2SO4溶液,右侧为0.5 mol·L-1MgSO4与2 mol·L-1H2SO4溶液的体系,在低温25℃和高温45℃下,加入1 V电场后透过侧VO2+浓度随时间的变化情况。温度对电场影响VO2+在膜中传质的情况也可以从图11中看出来,在协同场中,25℃时电场影响因子St为1.44~1.46,当温度升高为45℃时,St为1.54~1.58,并且随着时间的增加,这种电场加剧透膜传质的程度越发严重,温度越高,这一趋势越明显。
3.5 强制对流对协同场下膜中传质过程的影响
水力学条件是通过改变边界层厚度从而影响膜与溶液的接触面的离子的浓度和组成,进而影响离子跨膜渗透传质过程。若增加溶液的扰动,例如本实验中对钒电池溶液进行机械搅拌,则这种强制对流条件下形成的膜与溶液界面的滞流层相对于无搅拌的静止状态厚度降低,更加有利于离子的质量传递过程。图12为常温25℃下不同对流状态下0.5 mol·L-1VO2++2 mol·L-1H2SO4体系渗透侧 VO2+的浓度随时间的变化关系,可以发现对流状态下透膜传质现象更加严重。这也可以用St随时间变化图定量表征,如图13所示。协同场中,对流状态下的St在实验时间下由1.465增加到1.495,而静止状态下,St的变化范围是 1.445~1.462。虽然对流加剧了协同场下钒离子的跨膜传质过程,但是,对流作用降低了膜与溶液界面的滞流边界层,这有助于H+的传递过程,抑制了电池浓差极化,有助于提高电池的能量效率。所以,钒电池在实际应用中,往往刻意设计外循环从而造成一种电解液对流环境以提高电池性能。
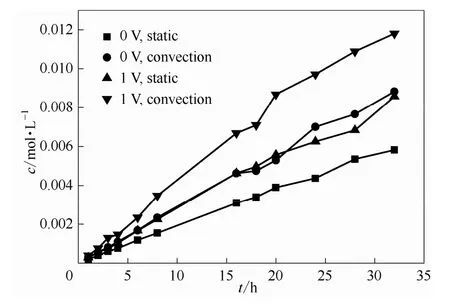
图12 不同对流状态下0.5 mol·L-1VO2++2 mol·L-1H2SO4体系渗透侧VO2+浓度随时间的变化关系Fig.12 Relationship between VO2+concentration with time in osmotic side of 0.5 mol·L-1VO2++2 mol·L-1H2SO4system in different convection condition
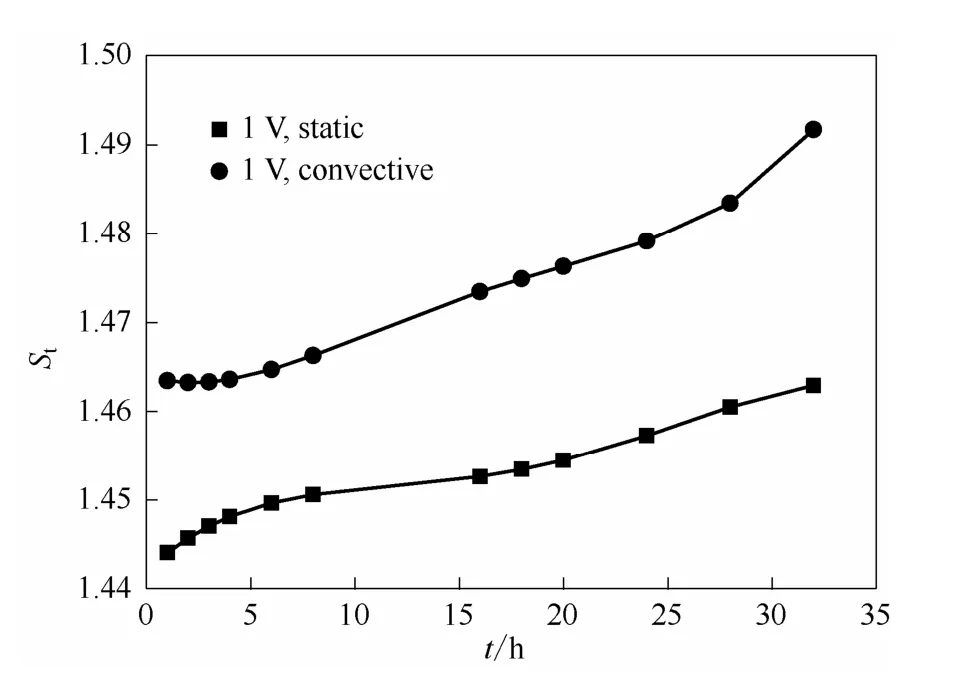
图13 不同对流状态下0.5 mol·L-1VO2++2 mol·L-1H2SO4体系电场因子随时间的变化关系Fig.13 Relationship between electric factor with time of 0.5 mol·L-1VO2++2 mol·L-1H2SO4system in different convection condition
3.6 对流和温度综合因素对协同场下膜中传质过程的影响
本实验还通过研究电池在高温和搅拌情况下VO2+跨膜渗透过程,考察了对流因素和高温因素对协同场中VO2+在Nafion膜中传质过程的综合影响。
图14表示了左侧为0.5 mol·L-1VOSO4与2 mol·L-1H2SO4溶液体系,右侧为 0.5 mol·L-1MgSO4与2 mol·L-1H2SO4溶液体系,在加入电场前后渗透侧VO2+浓度随时间的变化趋势。从图中可以看出,在温度为45℃搅拌转速为250 r·min-1的对流状态时,渗透侧 VO2+浓度相比于低温 25℃且电解液体系静止的情形均要大得多。
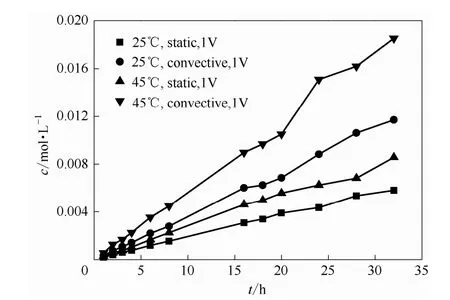
图14 不同温度和对流状态下0.5 mol·L-1VO2++2 mol·L-1H2SO4体系渗透侧VO2+浓度随时间的变化关系Fig.14 Relationship between VO2+concentration with time in osmotic side of 0.5 mol·L-1VO2++2 mol·L-1H2SO4system indifferent temperature and convection condition
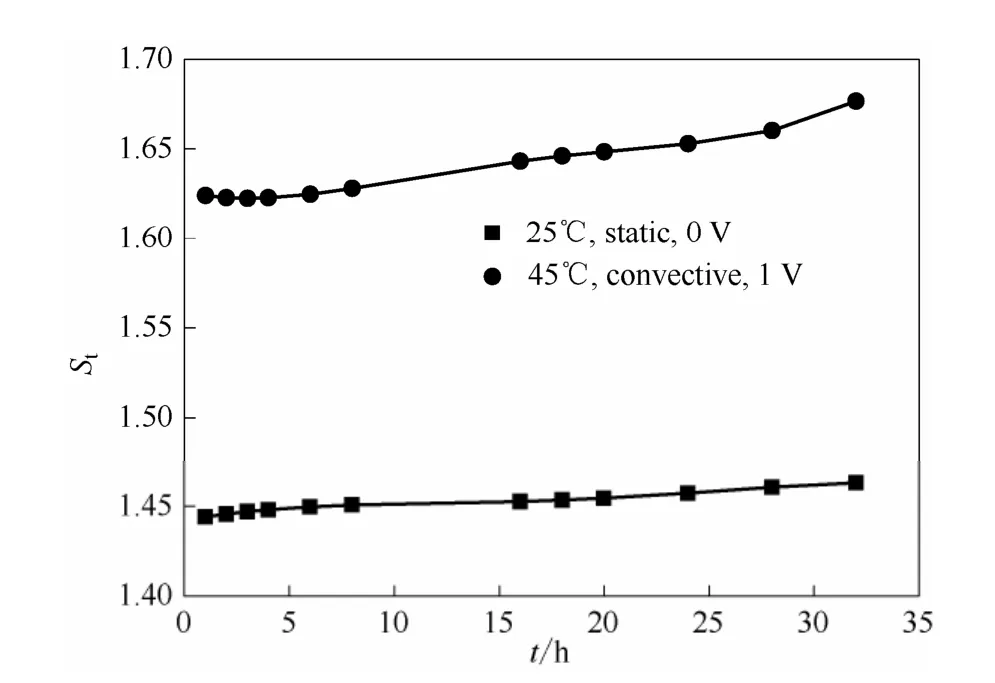
图15 不同温度和对流状态下0.5 mol·L-1VO2++2 mol·L-1H2SO4体系电场因子随时间的变化关系Fig.15 Relationship between electric factor with time of 0.5 mol·L-1VO2++2 mol·L-1H2SO4system in different temperature and convection condition
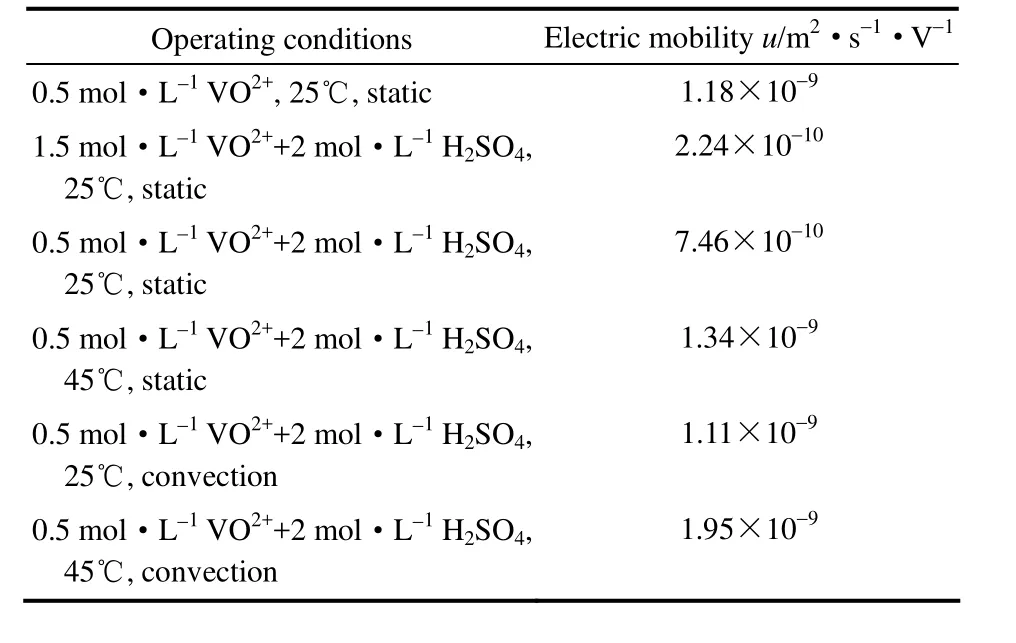
表3 各工况下VO2+电迁移率计算值Table 3 Calculated value of electric mobility of VO2+in different condition
图15直观反映了在同样施加大小为1 V的电压,高温对流状态时电场因子St相比于低温静止时要大得多,在45℃并且有对流存在时,St值在实验时间范围内从1.625增加到1.655,而25℃静止状态的St的范围为1.442~1.474,这表明:在协同场下高温对流能明显加剧钒离子的跨膜传质过程。
3.7 各工况下 VO2+在 Nafion117膜中表观电迁移率计算值
由实验所得数据,根据式(7)拟合出VO2+在Nafion117膜中的表观电迁移率,见表3。
4 结 论
通过研究协同场下VO2+在Nafion117膜中的传递过程,可知:外加电压越大,电场因子St越大。在各不同电压下,电场因子St均随着时间的增加而增大,且随着外电压的加大,St随时间增大的速度越来越快;反向电场在抑制离子跨膜渗透防止两极交叉污染方面有一定的应用价值;高浓度电解液在真实运行中的电场环境中,St明显偏小,有助于降低正负极交叉污染现象;在协同场中温度的提高使得St增加,加剧了钒离子在膜中的传质过程,并且随着时间的增加,这种电场加剧透膜传质的程度越发严重,温度越高,这一趋势越明显;电解液在对流状态下相比于静止状态,St值增加,电场影响传质过程更加明显。
符号说明
A——膜面积,m2
c——浓度,mol·L-1
D——扩散系数,cm2·s-1
E——电场强度,V·m-1
F——法拉第常数,C·mol-1
i——电流密度,mA·cm-2
j——传质通量,mol·m-3·s-1
L——正负极平行电极板间距,m
l——膜厚度,μm
St——电场因子
t——时间,s
U——电压,V
u——电迁移率,m2·s-1·V-1
V——电解液体积,L
z——电荷数
Γ——迁移数
下角标
d——扩散传质性质
e——电迁移性质
0 ——初始性质
1 ——钒液侧(左侧)性质
2 ——透过侧(右侧)性质
[1] Ludwig J, Juergen G, Fabjan C,et al. Possible use of vanadium redox flow batteries for energy storage in small grids and stand-alone photovoltaic systems [J].J. Power Sources, 2004, 127(1/2):98-104.
[2] Baker J. New technology and possible advances in energy storage [J].Energy Policy, 2008, 36(12):4368-4373.
[3] Jia Zhijun (贾志军), Song Shiqiang (宋世强), Wang Baoguo (王保国). A critical review on redox flow batteries for electrical energy storage applications [J].Energy Storage Science and Technology(储能科学与技术), 2012, 1(1):50-57.
[4] Rychcik M, Skyllas-Kazacos M. Characteristics of a new all-vanadium redox flow battery [J].Power Sources, 1988, 22(1):59-67.
[5] Huang K L, Li X G, Liu Su Q,et al. Research progress of vanadium redox flow battery for energy storage in China [J].Renewable Energy,2008, 33(2):186-192.
[6] Bae C H, Roberts E P L, Dryfer R A W. Chromium redox couples for application to redox flow batteries [J].Electrochim Acta, 2002, 48(3):279-287.
[7] Vijayakumar M, Sarah D Burton, Cheng Huang,et al. Nuclear magnetic resonance studies on vanadium(Ⅳ) electrolyte solutions for vanadium redox flow battery [J].Journal of Power Sources, 2010,195:7709-7717.
[8] Li Mengnan (李梦楠), Xie Xiaofeng (谢晓峰) , Yang Chun (杨春),et al. Characteristics of BMIMBF4as anode electrolyte additive for vanadium redox flow battery [J].CIESC Journal(化工学报), 2011,62(S2):135-139.
[9] Yang Chun (杨春), Wang Shubo (王树博), Xie Xiaofeng (谢晓峰) ,et al. Performance influence of hydroxyl radical on graphite felt electrode used in allvanadium redox flow battery [J].CIESC Journal(化工学报), 2012, 63(S1):188-193.
[10] Liu Ran (刘然), Liao Xiaoyan (廖孝艳), Yang Chun (杨春),et al.Different treatments of graphite electrode materials for vanadium redox flow battery [J].Chemical Industry and Engineering Progress(化工进展), 2011, 30:762-766.
[11] Ding C, Zhang H, Li X,et al. Morphology and electrochemical properties of perfluoro sulfonic acid ionomers for vanadium flow battery applications:effect of side-chain length [J].Chem Sus Chem,2013, 6:1262-1269.
[12] Yang Chun (杨春), Wang Jinhai (王金海), Xie Xiaofeng (谢晓峰),et al. AC impedance of influence of glycerin on all vanadium redox flow battery anodic electrolyte [J].CIESC Journal(化工学报), 2011,62(S1):163-167.
[13] Zhao Chengming (赵成明), Xie Xiaofeng (谢晓峰). Calculation of solvation free energies of VO V/VO2+ions in aqueous solution by using cluster-continuum model [J].CIESC Journal(化工学报), 2012,63(S2):132-135.
[14] Wang Wenpin (王文嫔), Wang Shubo (王树博), Xie Xiaofeng (谢晓峰),et al. Nonfluorinated anion exchange membranes based on imidazolium salts for all-vanadium redox flow battery [J].CIESC Journal(化工学报), 2013, 64(S1):203-208.
[15] Dong J Y, Zhang H M, Chen J. A simple model for the vanadium redox battery [J].Electrochimical Acta, 2009, 54:6827-6836.
[16] Al-Fetlawi H, Shah A A, Walsh F C. Non-isothermal modelling of the all-vanadium redox flow battery [J].Electrochimical Acta, 2009, 55:78-89.
[17] Zheng Q, Zhang H M, Xing F, Ma X K, Li X F, Ning G L. A three-dimensional model for thermal analysis in a vanadium flow battery [J].Applied Energy, 2014, 113:1675-1685.
[18] Wang Baoguo(王保国). Review of the development of proton exchange membranes in the renewable energy technology [J].Membrane Science and Technology(膜科学与技术), 2010, 30(1):1-8.
[19] Yin Haitao(尹海涛), Wang Baoguo(王保国). Influence of membrane diffusivity on the performance of a single vanadium flow battery [J].Battery Bimonthly(电池), 2006, 36(1):60-61.
[20] Lü Zhengzhong(吕正中), Hu Songlin(胡嵩麟) ,Luo Xuanli(罗绚丽) ,et al. Influence of proton exchange membrane on the performance of vanadium redox flow battery [J].Chemical Journal of Chinese Universities(高等学校化学学报), 2007, 28(1):145-148.
[21] Chen Jinqing(陈金庆), Lü Hongling(吕宏凌), Wang Baoguo(王保国) ,Yin Haitao(尹海涛). Study on the process of vanadium ions across membrane based on adsorption-diffusion mechanism [J].Contemporary Chemical Industry(当代化工), 2011, 40(9):893-895.
[22] Sun Hong (孙红), Luan Lihua(栾丽华), Wu Tiejun(吴铁军), Tang Yulan(唐玉兰), Wang Xun(王逊). Mass transfer in proton exchange membrane [J].Journal of Engineering Thermophysics(工程热物理学报),2012, 33(2):255-258.
[23] Kim Soowhan, Yan Jingling, Schwenzer Birgit, Zhang Jianlu, Li Liyu,Liu Jun, Yang Zhenguo (Gary), Hickner M A. Investigation of sulfonated poly(phenylsulfone) membrane for vanadium redox flow batteries[J].Electrochem. Comm., 2010 , 12(11) :1650-1653.
[24] Li Wenyue, Liu Jianguo, Yan Chuanwei. Multi-walled carbon nanotubes used as an electrode reaction catalyst for VVO2+for a vanadium redox flow battery [J].Carbon, 2011, 49 (11):3463-3470.
[25] Tan Ning(谭宁), Huang Kelong(黄可龙),Liu Suqin(刘素琴). Studies on ion exchange membrane performance in vanadium solution for all vanadium flow battery [J].Chinese Journal of Power(电源技术),2004, 28(12):775-778.
