高压下硝基甲烷分子温度的密度泛函计算
2015-03-23董光兴葛素红李德华
董光兴, 葛素红,李德华
(1.河西学院物理与机电工程学院, 张掖 734000; 2. 中国科学院武汉物理与数学研究所波谱与原子分子物理国家重点实验室, 武汉 430071; 3. 四川师范大学物理与电子工程学院, 成都610066 )
高压下硝基甲烷分子温度的密度泛函计算
董光兴1, 葛素红1,2,李德华3
(1.河西学院物理与机电工程学院, 张掖 734000; 2. 中国科学院武汉物理与数学研究所波谱与原子分子物理国家重点实验室, 武汉 430071; 3. 四川师范大学物理与电子工程学院, 成都610066 )
用密度泛函理论在B3LYP/6-311++G(2d,2P)计算水平上对硝基甲烷分子进行了结构优化、 频率和热化学分析. 发现: 在相同温度条件下改变压强, 分子熵函数产生了改变, 当温度和压强条件相同时, 对于不同物质熵函数的改变是相同的. 以热力学理论中麦克斯韦关系为基础, 通过计算等温过程中分子的熵函数对压强的变化率, 用数值拟合方法得到不同压强条件下分子温度的表达式:T=T0+(1-B)[18.3858+0.5392P]V0, 式中T0、 V0分别表示分子系统初态的温度和体积, T、 V分别表示系统在末态的温度和体积, B是体积的压缩比. 在选定参数的情况下该表达式可以计算不同压强条件下CHNO含能材料的分子温度. 同时, 以硝基甲烷为验证, 选取基本参数V0和B, 计算其在C-J条件对应的爆压14GPa下, 分子温度为3461K, 对应爱因斯坦温度, 相当于3228cm-1的能量, 在实验中该能量足以激发硝基甲烷分子内振动能量重新分配过程, 有可能激发C-N键的红外振动而引起单分子分解反应的发生. 因此, 此表达式可用于预测含能材料撞击点火过程单分子分解可能的反应通道.
热力学; 麦克斯韦关系; 多声子迁移模型; 含能材料; 硝基甲烷(NM)
1 引 言
大多数含能材料在常温下是固相, 也有一部分是液态. 液体在其受力作用时间很短的情况下表现出固体的脆性, 在一些具体的研究中可以将液体的含能材料作为瞬态的固体来处理. 含能材料撞击点火过程是指材料在一个极短的时间内受到外界一定阈值的撞击从而发生爆炸的过程, 因此, 在对由有机分子组成的单相的含能材料撞击点火的研究中, 可以将单质的材料看成分子晶体. 对于它们撞击点火过程研究的出发点主要有两个方面, 其一, 从基本物理过程的角度分析, 外界撞击在很短的时间内给固体材料施加了强大的压力, 导致材料内部热力学状态发生变化, 温度升高的同时内能增加, 继而发生爆炸. 其二, 从含能材料爆炸过程的化学性质来看, 它是以凝聚相和气相化学反应为基础的多组分和多阶段过程, 是由于材料内部温度升高而导致的分子的分解过程及后续的系列化学反应过程, 需要深入研究热分解的各基元反应历程, 建立反应物、 过渡态(包括中间体)和产物之间的联系[1]. 含能材料的同一个撞击点火过程同时包括了这两方面的物理化学的变化过程, 材料内部的压强和温度有急剧的变化, 点火过程中有作为分子解体的第一步反应的吸热反应和接下来一系列的放热反应, 这就使得研究材料温度和压强的阈值条件对撞击点火过程的微观机理有非常重要的意义.
在含能材料领域内, 基于热力学基本理论和物态方程的建立对含能金属材料冲击压缩特性的实验和理论计算已有很多研究[2-7]. 而对由CHNO有机分子组成的含能材料在高压下的理论和实验的研究比较有限. Perger W F用密度泛函方法计算了4Gpa以内太安炸药晶体在高压下的晶格参数和内坐标[8]. 逯来玉等对季戊四醇分子晶体在高温下的结构和振动性质进行计算研究[9,10]. 他们的研究发现除了极少数, 如C-C键的伸缩振动模式是随温度的升高有缓慢的升高, 大多数晶体分子内振动的频率随温度的升高都有比较平缓的降低, 他们认为这说明分子内的键强随着温度的升高存在变弱的趋势. 在分析分子晶体含能材料在撞击点火过程中材料分解的微观机理时, 这样的研究结论有非常重要的意义, 因为固体材料的解体必须首先发生在材料分子内部, 是由分子的某一个化学键首先离解之后而导致系列反应的发生的.
硝基甲烷(NM)是一个非常典型且分子较小的由CHNO组成的硝基复合物, 它可以用于制造炸药, 还可用作火箭的液体燃料. 作为分子结构较为简单的含能材料, 对它的研究能够作为其他含能大分子体系, 尤其是由20-50个原子组成的分子系统的基准. 因此, 在正常和极端的热力学条件下的对于硝基甲烷材料的理论和实验的研究是非常广泛的. 早在1959年Hubbard和Johnson理论地计算了NM爆炸过程的热力学参数, 他们的计算结果与实验的结果吻合很好[11,12]. 2003年Manaa对硝基甲烷在均匀受压和轴向受压进行了量子化学计算[13], 研究认为当体积压缩至原来的一半时, 压强上升至50GPa, 最高占据轨道和最低空轨道的能级间隔减小了约0.6eV, 但是, 最高占据轨道和最低空轨道能级同时升高, 其差值却几乎随体积的变化单调下降, 带隙减小从理论上可以认为感度在增加. 2011年Han对硝基甲烷在高温过程进行了模拟计算[14], 认为硝基甲烷在300K时分解起始步骤为分子间质子迁移, 生成CH3NOOH和CH2NO2. 在较低的温度(2500~2000K)第一步反应则为异构化生成CH3ONO的过程. 同时在反应刚开始极短时间内可伴随分子内质子迁移生成CH3NOOH. 而对于硝基甲烷分子的分解机理的研究则有很多报道[15-21]. 对它的光解研究表明, 气态情况下分子沿C-N键的均裂主要生成甲基和二氧化氮[18]. 电子碰撞光谱研究报道了硝基甲烷分子的最低单态到三态的激发跃迁[19]. 有作者在实验中观测分析认为在硝基甲烷光解为甲基和二氧化氮的过程中, 分子态跃迁起着很大的作用, 他们认为合理的反应机理应该是分子先激发为三态然后再分解为甲基和二氧化氮的[20].
但是, 从整个撞击点火反应过程来看, 首先是外界的机械撞击引起了材料的压强和温度等物态参数的改变, 其次是材料分子的某个化学键受到激发而发生断裂, 引起了整个的系列反应. 撞击机械能引起的材料内部温度和压强条件的变化是材料分子分解反应发生的先决条件, 这就需要在研究由CHNO有机分子组成的含能材料撞击点火机理时考虑外界撞击对材料内部产生的影响, 将材料的热力学状态变化和材料单分子分解条件相互联系. 因此, 本文选取具有代表性的含能材料---硝基甲烷为研究对象, 以热力学理论为基础, 在密度泛函算法基础上推导了含能材料分子高压下的温度压强关系式, 并借鉴多声子迁移模型的基本理论预测了硝基甲烷在撞击诱导点火过程的基元反应.
2 理论模型和计算方法
2.1 多声子迁移模型的基本思想
在撞击点火过程中外界冲击和撞击产生的机械能是被转移到材料的分子反应中心的, 这说明撞击点火过程是由两个不同的过程组成的. 多声子迁移模型认为这说明外界撞击产生的机械能是经过在固体中激发声子, 再经声子将能量传递转移至分子键, 激发分子键的红外振动, 导致分子键的断裂, 激发随后而来的放热化学反应, 导致区域温度急速上升, 热点形成, 材料迅速点火爆炸[22-25]. 晶体材料的声子振动模式频率较低, 而对应分子红外振动频率却分布较广. 多声子迁移模型认为, 被机械撞击激发的声子经双声子吸收过程, 先激发分子中较低的红外振动模式, 将这种低频红外振动模式称为门模式, 门模式的频率一般在(200,700)cm-1. 然后, 门振动模式作为低频振动模式, 可与声子耦合. 当能量持续从过热声子区流入到门振动, 经过多声子迁移, 高一级的分子振动模式会通过分子内振动能量的迁移而被门模式所激发, 最终达到分子内振动能量的某一阈值, 然后, 形成了分子红外振动能量在分子内的重新分配过程, 这种振动态的迁移一直持续到有足够的能量被定域到导致反应的分子反应路径上, 导致键的裂解, 分子解体, 第一步吸热反应发生, 接下来会在局部发生一系列的放热反应, 导致温度升高, 热点形成, 点火完成[23]. 硝基甲烷分子振动能量重新分配过程的实验和理论模拟都有研究报道[26,27]. 实验中首先激发了硝基甲烷分子C-H键的红外振动(~3000cm-1), 发现分子的红外振动迁移主要由三个过程组成, 先是被激发的高能量红外振动在几个皮秒内振动冷却, 激发了化学键的能量较低的红外振动, 范围大致为(~1600-1400cm-1). 然后能量较低的这些振动发生弛豫过程, 激发了化学键的能量更低的红外振动模式, 范围大致为(~1100-480cm-1). 最后, 被激发的这些红外振动模式发生振动冷却过程, 从而激发声子振动模式[26].
因此, 在撞击点火过程中, 当存在足够的能量以激发材料分子能量较高的红外振动模式, 且达到分子内振动能量重新分配的阈值, 则会有这样阶梯式的分子红外振动能量迁移. 当作为分子分解的化学键的红外振动被激发, 就会造成该化学键的断裂, 单分子分解反应发生, 引发接下来的点火爆炸过程.
2.2 高压下分子温度的预测和计算原理
到目前为止, 虽然多声子迁移模型是一个比较完美的理论, 用它来解释含能材料的撞击点火过程是合乎逻辑的. 但是, 由于声子振动模在实验上难以检测和验证, 多声子迁移模型理论被认为缺乏直接的实验依据[28]. 无论如何, 含能材料的撞击点火过程是将撞击机械能传递到分子内部而导致分子的解体的, 多声子迁移模型认为机械能转化为声子振动的能量后迁移至分子内部的振动能, 从能量守恒的角度分析, 也可以认为是撞击机械能转化为热能而后迁移至分子内部激发分子内部化学键的红外振动.
对于多原子体系由N个单原子组成的分子本身, 或者由M个分子组成的晶体原胞都可以看成是宏观热力学系统. 则系统的热力学状态函数的改变只取决于状态而与过程无关, 依据热力学基本理论麦克斯韦关系式[29]:
(1)
等压过程中体积随温度的变化率等于等温过程状态函数熵对压强的偏导数的负值. 若将撞击点火过程中系统温度和压强的急剧变化过程看成是一系列等压过程或者等温过程的积累, 在撞击点火过程中分子系统内部的温度压强关系可以通过计算熵函数的改变而获得:
(2)
在一无限小等压过程中, 可将偏导数表示为物理近似关系式:
(3)
由(3)式可以获得系统的温度压强关系式:
(4)
式中T0、V0分别表示分子系统初态的温度和体积,T、V分别表示系统在末态的温度和体积, B是体积的压缩比, 在正常情况下, 要求其小于1, 定义为B=V/V0.

(5)

由于第一性原理的密度泛函理论(DFT)方法系统误差要比HF方法小[31], 且密度泛函B3LYP杂化方法, 不但对交换能做密度梯度校正, 而且对相关能也做了梯度校正, 提高了密度泛函计算精确度[32]. 本文采用密度泛函理论方法, 在B3LYP/6-311++(2d,2P)计算水平上, 对硝基甲烷的结构进行了全优化, 并在同一水平上计算了298.15K温度、 不同压强下的热化学参数. 所有计算工作用Gaussian09程序包完成. 频率计算验证无虚频, 而且有六个几乎为零的频率, 说明计算研究是在最小势能面上进行的.

表1 硝基甲烷分子的几何参数
键长单位(Å);键角和二面角单位(o)
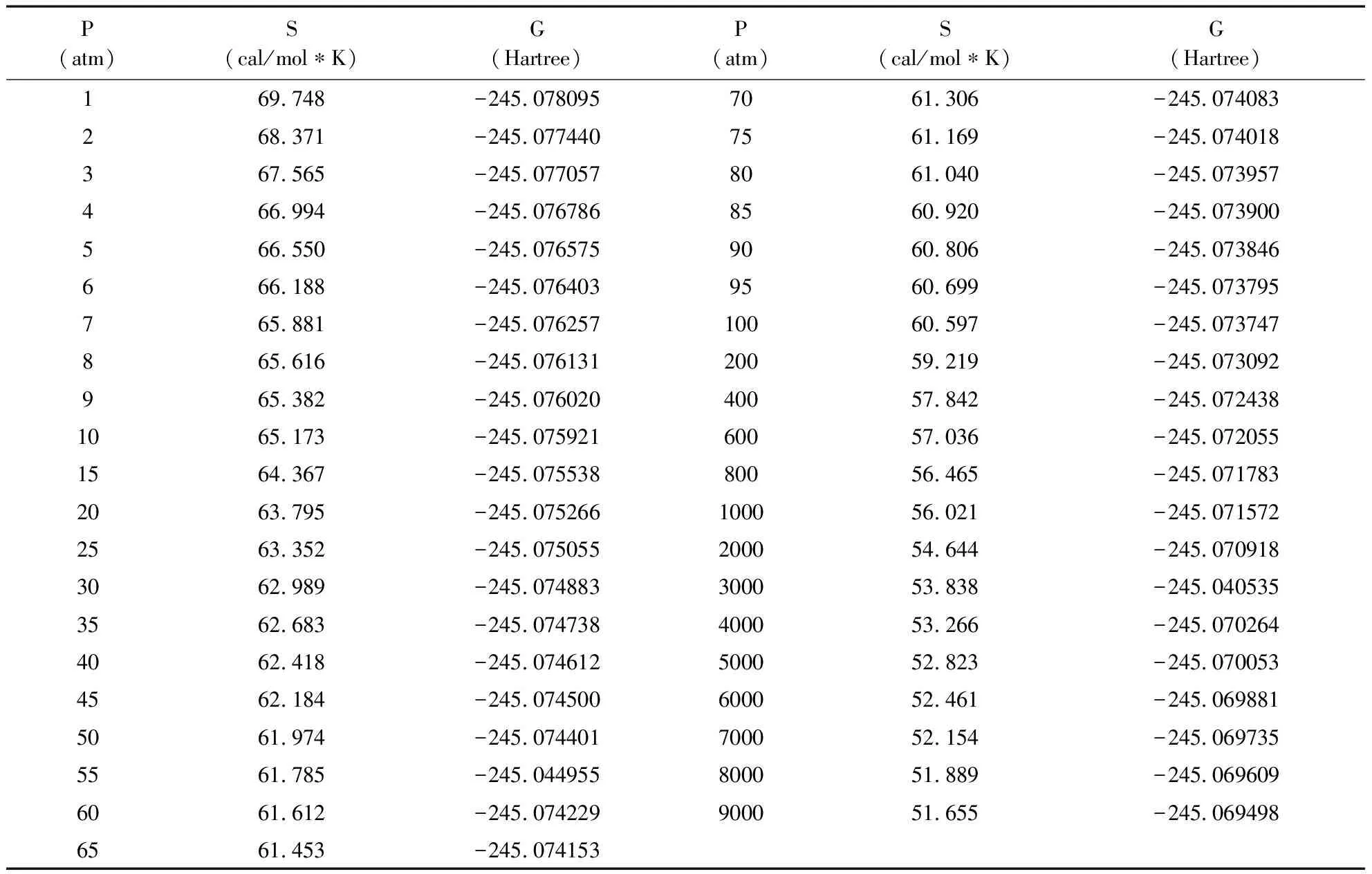
表2 不同压强下硝基甲烷分子的熵和自由能的计算值
3 计算结果与讨论
3.1 硝基甲烷单分子温度的模拟公式

图1 等温过程分子熵随压强变化率与压强的关系(1-100)atmFig.1 The molecular entropy change rate with pressure versus pressure of NM in (1-100)atm
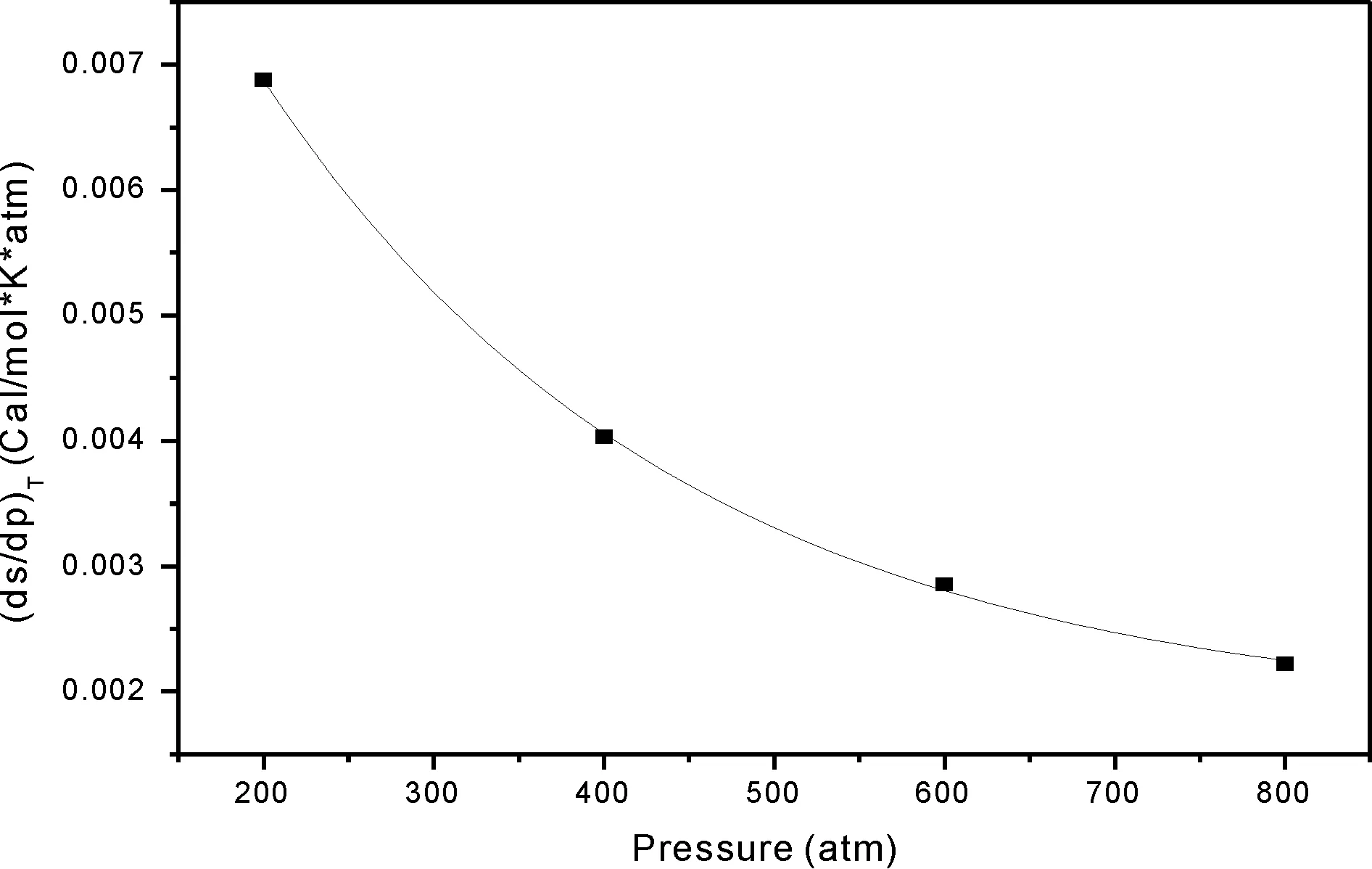
图2 等温过程分子熵随压强变化率与压强的关系(100-1000)atmFig.2 The molecular entropy change rate with pressure versus pressure of NM in (100-1000)atm
首先运用密度泛函理论中的B3LYP/6-311++G(2d,2P)方法对基态的硝基甲烷分子进行几何优化, 得到稳定的几何构型, 确保计算是在势能面的最低点进行. 分子的几何构型参数列在表1中. 根据文献, 基态的硝基甲烷分子稳定构型参数是:C-H键键长为1.088 Å, C-N键键长为1.489 Å, N-O键键长为1.224 Å, 键角∠NCH为107°, ∠ONO为125.3°[33]. 表1中基态分子的几何构型参数与其实验值相比较, 键长的最大偏差为0.005Å, 键角的误差基本是1°, 这样小的误差说明分子的构型优化是比较成功的, 在这个基础上再进行频率分析计算就会得到更可靠的结果. 然后, 保持温度在298.15K的条件, 改变压强参数, 在B3LYP/6-311++G(2d,2P)计算这个构型下的硝基甲烷分子的热化学参数, 计算发现分子的熵函数和自由能(经过零点修正)的数值随压强的改变而改变. 计算数据在表2中列出.
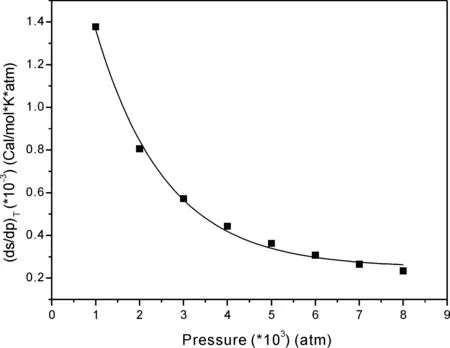
图3 等温过程分子熵随压强变化率与压强的关系(1000-8000)atmFig.3 The molecular entropy change rate with pressure versus pressure of NM in (1000-8000)atm

图4 等压过程分子温度随体积的变化率与压强的关系图线Fig.4 The molecular temperature change rate with volume versus pressure of NM
根据表2数据计算等温过程分子体系的熵函数随压强的变化率并拟合变化率与压强的关系图形, 按压强分区绘制如图1、 图2和图3所示. 根据表2数据, 依据公式(2)计算不同压强条件下温度随体积的变化率, 拟合得到温度对体积的变化率与压强呈线性, 如图4所示, 其相关系数R为0.99927,R2等于0.9985, 这个数据说明数据的线性相关性较好.拟合所得方程为:
(6)
拟合方程(6)给出了硝基甲烷材料分子在等压过程中温度对体积的变化率.将(6)式代入(4)式可得最终的拟合推导结果, 即不同压强下分子温度的计算公式:
=T0+(1-B)[18.3858+0.5392P]V0
(7)

图5 硝基甲烷分子结构图Fig.5 Optimized geometrical configurations of NM
3.2 不同条件下硝基甲烷分子分解通道分析
常温下硝基甲烷是液体状态, 人们没有获得它的撞击感度实验值. 一般情况下人们将含能材料爆轰时所产生的爆轰波阵面的压力称为爆压, 也称为C-J压力.实验上可以得到常用炸药材料的爆压为10-40GPa, 硝基甲烷的爆轰压力可达到14GPa[34]. 传统的固体含能材料热力学爆炸理论是有名的Chapman-Jouguet模型[35], 这个模型认为C-J温度和C-J压力就是含能材料的爆轰条件. 因此选取14GPa压强条件作为硝基甲烷的点火爆轰条件.
图5所示为硝基甲烷分子结构图, 考虑电子云分布, 微观上将单个分子看成球型结构, C-N键中点为球心, C-N键长的一半加上N—O键和C-H键中最长的数值可以认为是球的半径, 由此估算得硝基甲烷分子的体积为6π×10-30m3. 由于撞击点火过程中含能材料受到猛烈的外界撞击, 外界撞击的力远大于分子间力的作用, 因此, 对于结构疏松的分子晶体, 忽略分子间力的作用,将未受撞击作用时硝基甲烷分子的体积作为初态体积, 则有V0=6π×10-30m3, 在撞击发生之前材料与环境达到热平衡, 选取分子的初始温度T0等于环境温度298.15K. 同样,在这种近似条件下, 可将疏松结构的分子晶体在撞击前后的体积的压缩比B值选取为0.793[23]. 依据(7)式可计算得在压强达到14GPa时, 硝基甲烷分子的温度值为:3461K, 这样的分子温度相当于0.4eV的能量, 对应爱因斯坦温度, 相当于3228cm-1的能量.

表3 硝基甲烷分子的振动频率
a本文理论计算的频率值;b实验文献[26]中的频率值
硝基甲烷分子C-H键的红外振动能量就在3000cm-1左右, 如表3所示是硝基甲烷分子的红外振动光谱理论计算值和实验值. 按照Deak等人的实验结果, 在激发了硝基甲烷分子C-H键的红外振动(~3000cm-1), 之后, 发现激发的高能量红外振动在几个皮秒内振动冷却,激发了化学键的能量较低的红外振动, 范围大致为(~1600-1400cm-1), 然后能量较低的这些振动发生弛豫过程, 激发了化学键的能量更低的红外振动模式, 范围大致为(~1100-480cm-1). 最后, 被激发的这些红外振动模式发生振动冷却过程, 从而激发声子振动模式[26]. 那么, 当外界撞击导致材料分子所受压强达到14GPa时, 分子温度所对应的能量相当于3228cm-1, 足以激发硝基甲烷分子C-H键的红外振动, 这将导致分子振动能量重新分配过程的发生, 实验显示, 这个过程会激发化学键的能量较低的红外振动, 其范围分别为(~1600-1400cm-1)和(~1100-480cm-1). 当CN键的伸缩振动模式918(1379)cm-1被激发后, 硝基甲烷分子就有可能沿着这个反应坐标分解产生分解反应, 导致材料分子解体, 引发接下来的一系列链式反应, 使点火反应发生:
CH3NO2→CH3+NO2
(8)
因此, 可以认为本文的理论计算公式与硝基甲烷分子振动能量重新分配过程的实验和理论模拟的研究结果相一致[26,27]. 可以认为14GPa就是硝基甲烷材料C-J条件. 在本文的近似条件下, 选取相同的参数V0和B, 根据公式(7)还可以计算不同压强条件下的分子温度和其对应的振动能量, 计算结果在表4中列出. 从表4数据可以看出当压强条件达到12GPa时, 分子温度对应的能量数值已经达到了实验中需要激发分子C-H键红外振动的能量, 但材料实验中硝基甲烷的爆压却是14GPa. 造成这一偏差的原因在于热能的传递和利用有效率的问题, 在被有效利用的过程中热能总是存在着散失. 因此, 在表4的计算中选取数据时, 计算值都要大于实验给出的数值, 由此来推断在对应压强下可能被激发的分子的红外振动模式, 这正是推断晶体材料单分子分解情况的前提.
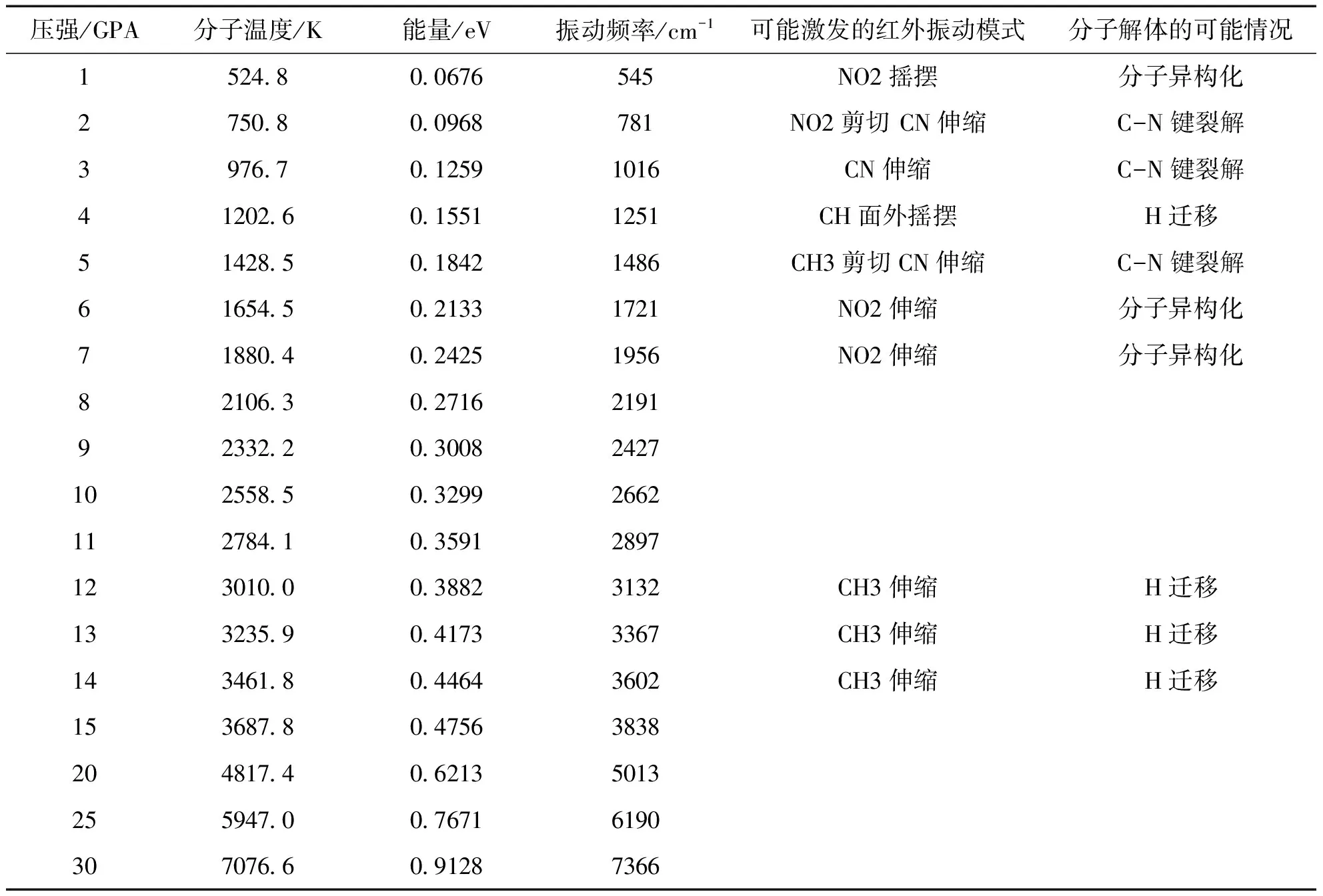
表4 不同压强下硝基甲烷分子温度和能量
就目前的实验事实发现, 在激发了硝基甲烷分子C-H键的红外振动之后, 分子中发生了振动能量的重新分配过程, 使得CN键的伸缩振动模式或者其他的红外振动模式有可能被激发, 而导致单分子分解反应的发生. 但是, 实验并没有给出这样一种情形, 那就是直接激发分子的C-H键的红外振动模式或者其他的红外振动模式之后, 会不会存在晶体材料单分子分解反应的发生. 因此, 在表4中, 只有当压强达到或者大于14GPa, 才可以确定地认为从本文的理论预测和实验两方面都能判断分子的CN键的伸缩振动模式可能被激发, 这个条件下会有分子分解反应的发生, 反应路径沿着C-N键. 当压强达到3GPa时, 分子温度所提供的能量就足够激发CN键的伸缩振动模式了, 在其他小于14GPa的压强条件下, 分子温度所提供的能量也足够激发另外的红外振动模式, 分子解体的可能情况在表4中可以对应分析, 但是反应发生的可能性无从判断. 对于这样的情形, 需要实验研究给出证明.
4 结 论
撞击点火过程是指含能材料受到猛烈的外界撞击, 从而导致材料的点火反应发生引起爆炸的过程. 由于外界撞击的力远大于分子间的相互作用, 因此, 对于结构疏松的分子晶体, 忽略分子间力的作用. 本文基于这样的近似处理, 以热力学基本理论为基础, 将材料的撞击点火过程看成系列等压过程, 通过计算多粒子的分子体系熵函数的改变推导拟合得到一个随压强而变化的分子温度函数关系式, 进而计算不同压强条件下材料分子的温度. 在爱因斯坦特征温度的概念下, 将分子温度换算成能量数值, 配合现存的实验结论, 由此判断该温度阈值是否足够激发分子红外振动正则模式, 并在分子内部形成振动能量的重新分配过程. 本文选取硝基甲烷为研究分子, 计算并拟合出高压下分子温度公式, 选择材料的初状态和分子的基本参数, 计算得到硝基甲烷爆压条件14GPa下的分子温度为3461K, 这样的分子温度相当于0.4eV的能量, 对应爱因斯坦温度, 相当于3228cm-1的能量.对比DeaK等人的实验研究, 该能量正好可以激发硝基甲烷分子C-H键的红外振动模式而导致分子内振动能量重新分配过程, 这样的分配过程最终会激发C-N键的红外振动而引起单分子分解反应的发生. 这说明本文拟合的理论公式计算的结果与硝基甲烷材料的爆轰条件相符合, 可用于预测计算CHNO含能材料在高压下的分子温度, 或者计算预测含能材料产生点火反应的压强条件, 在有关于分子振动能量重新分配过程的实验结果支持的条件下, 本文的计算结果也可用于预测外界撞击条件下单分子分解反应的可能路径和通道.
[1]JuXH,YeCC,XuSY.Overviewonquantumchemicalcomputingandmoleculardynamicsimulationsofenergeticmaterials[J].ChineseJournalofExplosives&Propellant, 2012, 35(2): 1 (in Chinese)[ 居学海, 叶财超, 徐司雨.含能材料的量子化学计算与分子动力学模拟综述[J]. 火炸药学报, 2012, 35(2): 1]
[2] Shi A S, Zhang X F, Qiao L,etal. Theoretical calculation on shock compression characteristics of multifunctional energetic structural material[J].ExplosionandShockWaves, 2013, 33(2): 148(in Chinese) [史安顺, 张先锋, 乔良, 等. 多功能含能结构材料冲击压缩特性的理论计算[J]. 爆炸与冲击, 2013, 33(2): 148]
[3] Zhang X F, Qiao L, Shi A S. A cold energy mixture theory for the equation of state in solid and porous metal mixtures[J].JournalofAppliedPhysics, 2011, 110(1): 013506.
[4] Wu Q, Jing F Q. Unified thermodynamic equation-of-state for porous materials in a wide pressure range [J].JournalofAppliedPhysics, 1995, 57(1): 49.
[5] Jordan J L, Ferranti L, Austin R. Equation of state of aluminum-iron oxide-epoxy composite [J].JournalofAppliedPhysics, 2007, 101(9): 093520.
[6] Yuan X L, Wei D Q, Cheng Y,etal. Thermodynamics properties of Zr4Al2under high pressure from first-principle calculations[J].J.At.Mol.Phys., 2012, 29(2): 325 [苑晓丽, 魏冬青, 程艳, 等. Zr4Al2高压下热动力学性质的第一性原理研究[J]. 原子与分子物理学报, 2012, 29(2): 325]
[7] Wang J, Guo F, Cheng X L,etal. Reactive molecular dynamics simulations of initial decomposition of TATB under high temperature and high pressure[J].JournalofSichuanUniversity:NaturalScienceEdition, 2013, 50(3): 580(in Chinese) [王君, 郭峰, 程新路, 等. TATB高温高压下初始分解反应的分子动力学模拟[J].四川大学学报: 自然科学版, 2013, 50(3): 580]
[8] Perger W F, Vutukuri S, Dreger Z A,etal. First principles vibrational studies of pentaerythritol crystal under hydrostatic pressure[J].Chem.Phys.Lett., 2006, 422: 397.
[9] Lu L Y, Zhou X L, Ji G F,etal. Structural and vibrational properties underhigh temperature of solid pentaerythritol by molecular dynamics simuations[J].J.At.Mol.Phys., 2012, 29(5): 908 (in Chinese) [逯来玉, 周小林, 姬广富, 等. 高温季戊四醇晶体的结构和振动性质的分子动力学模拟[J]. 原子与分子物理学报, 2012, 29(5): 908]
[10] Lu L Y, Wei D Q, Chen X R,etal. First principle calculations of structures and electronic properties of solid pentaerythritol under pressure[J].Chin.Phys.Lett., 2008, 25: 3368.
[11] Hubbard H W, Johnson M H. Initiation of detomations [J].J.Appl.Phys., 1959, 30: 765.
[12] Campbell A W, Davis W C, Travis J R. Shock initiation of detonation in liquid explosives [J].Phys.Fluids., 1961, 4: 498.
[13] Manaa M R, Fried L E, Reed E J. Explosive chemistry: simulating the chemistry of energetic materials at extreme conditions [J].J.Comput.Aid.Mol.Des, 2003, 10(2): 75.
[14] Han S P, Van Duin A C T, Goddard Ⅲ W A,etal. Thermal decomposition of condensed-phase nitromethane from molecular dynamics from ReaxFF reactive dynamics [J].J.Phys.Chem. B, 2011, 115: 6534.
[15] Blais N C, Engelke R, Sheffield S A. Mass spectroscopic study of the chemical reaction zone in detonating liquid nitromethane [J].J.Phys.Chem. A, 1997, 101: 8285.
[16] Gruzdkov Y A, Gupta Y M. Emission and fluorescence spectroscopy to examine shock-induced decomposition in nitromethane [J].J.Phys.Chem. A, 1998, 102: 8325.
[17] Miller P J, Piermarini G J, Block S. An FT-IR micro-spectroscopic method for kinetic measurements at high temperatures and high pressures [J].AppliedSpectroscopy, 1984, 38: 680.
[18] Moss D B, Trentelmen K A, Houston P L. 193 nm photodissociation dynamics of nitromethane [J].J.Chem.Phys., 1992, 96: 237.
[19] Flicker W M, Mosher O A, Kuppermann A. Variable angle electron-impact excitation of nitromethane [J].J.Chem.Phys., 1980, 72: 2788.
[20] Honda K, Mikuni H, Takahasi M. Photolysis of nitromethane in gas phase at 313 nm[J].Bull.Chem.Soc.Jpn., 1972, 45: 3534.
[21] Liu H, Zhao J J, Wei D Q,etal. Structural and vibrational properties of solid nitromethane under high pressure by density functional theory [J].J.Chem.Phys., 2006, 124: 124501.
[22] Dlott D D. Multi-phonon up-pumping in energetic materials [J].Overviewsofrecentresearchonenergeticmaterials.Adv.Ser.Phys.Chem., 2005, 16: 303.
[23] Tokmakoff A, Fayer M D. Chemical reaction initiation and hot-spot formation in shocked energetic molecular materials [J].J.Phys.Chem., 1993, 97: 1901.
[24] Dlott D D, Fayer M D. Shocked molecular solids: vibrational up pumping, defect hot spot formation, and the onset of chemistry [J].J.Phys.Chem., 1990, 92: 3797.
[25] Dlott D D. Picosecond dynamics behind the shock front [J].LeJournaldephysiqueⅣ, 1995, 5(4): C4-337.
[26] Deak J C, Iwaki L K, Dlott D D. Vibrational energy redistribution in ployatomic liquids: ultrafast IR-Raman spectroscopy of nitromethane [J].J.Phys.Chem. A, 1999, 25: 971.
[27] Kabadi V N, Rice B M. Molecular dynamics simulations of normal mode vibrational energy transfer in liquid nitromethane [J]J.Phys.Chem. A, 2004, 108: 532.
[28] Zeman S. Sensitivities of high energy compounds [J].Struct.Bond., 2007, 12: 195.
[29] Wang Z C.Thethermodynamicstatisticalphysics[M]. Beijing: Higher Education Press, 2003(in Chinese)[汪志诚. 热力学统计物理学[M]. 北京: 高等教育出版社, 2003]
[30] Huang K.Solidstatephysics[M]. Beijing: Higher Education Press, 1988(in Chinese)[黄昆. 固体物理[M]. 北京: 高等教育出版社, 1988]
[31] Wiberg K B, Hadad C M, Lepage T J,etal. An analysis of the effect of electron correlation on charge density distribution [J].J.Chem.Phys., 1992, 96: 671.
[32] Joseph W, Ochterski Ph D.ThermochemistryinGaussian[M]. Gausssian Inc. 13, 2000.
[33] Huber K P, Herzberg G.Molecularspectraandmolecularstructure:constantsofdiatomicmolecules[M]. London: Van Nostrand Reinhold Co, 1979.
[34] Hardesty D R. An investigation of the shock initiation of liquid nitromethane [J].CombustionandFlame, 1976, 27: 229.
[35] Zeldovich Y B, RAizer Y P.Physicsofshockwavesandhightemperaturehydrodynamicphenomena[M]. New York: Academic Press, 1966.
Density functional theory study on the temperature of nitromethane molecule under pressure
DONG Guang-Xing1,GE Su-Hong1,2, LI De-Hua3
(1. Department of Physics, Hexi University, Zhangye 734000, China; 2. State Key Laboratory of Magnetic Resonance and Atomic and Molecular Physics, Wuhan Institute of Physics and Mathematics, Chinese Academy of Sciences, Wuhan 430071, China; 3. College of Physics and Electronic Engineering, Sichuan Normal University, Chengdu 610066, China)
In the present paper B3LYP method complied with the 6-311++G(2p,2d) basis set was employed to investigate the molecular property of nitromethane. Based on a systematic study of the molecular structure, electronic energy, normal vibration mode and thermal chemical functions in nitromethane via the lowest singlet, it is found that the decrease of entropy with pressure is evident if we keep the temperature constant. Based on Maxwell relations of the thermodynamic we deduced the fitting equation as follow:T=T0+(1-B)[18.3858+0.5392P]V0, which can calculate the temperature of the energetic molecules under different pressures. Further more, the temperature of the nitromethane under 14GPa is 3461K, which equates to 3228cm-1. This energy value could excite the C-H stretching and the intramolecular vibrational redistribution process, the process could generate the decomposition of the molecule. Thus, the fitting equation can expect the decomposed path way of the energetic molecules.
Thermodynamic; Maxwell relations; Multiponon transfer mode; Energetic Materials; Nitromethane
103969/j.issn.1000-0364.2015.02.002
2014-06-25
国家自然科学基金(41171175)
董光兴(1971—),男,甘肃平凉人,副教授,硕士,主要从事光学、量子力学等的教学科研工作.
葛素红.E-mail: gesuhong@163.com
O642
A
1000-0364(2015)02-0181-09
