均相催化O2氧化醇的研究进展(上)
2015-02-05王连月
王连月,高 爽
(中国科学院 大连化学物理研究所 洁净能源国家实验室,辽宁 大连 116023)
特约述评
均相催化O2氧化醇的研究进展(上)
王连月,高 爽
(中国科学院 大连化学物理研究所 洁净能源国家实验室,辽宁 大连 116023)
综述了以分子氧为氧源的催化氧化醇体系,包括过渡金属(Cu,Fe,Co,V,Ru,Pd,Au)催化体系、非金属催化体系(2,2,6,6-四甲基哌啶氮氧自由基)及2,3-二氯-5,6-二氰对苯醌等其他小分子体系。对各催化体系的适用范围和优缺点进行了分析,阐述了主要催化体系的催化机理,对已有研究成果进行了总结,并在此基础上对该领域的发展趋势作了展望。
分子氧;醇;选择氧化;均相催化
醇选择性氧化为醛或酮是有机合成中非常重要的官能团转化反应,无论对基础研究还是对大规模工业生产都具有十分重要的意义[1]。醛和酮是重要的工业原料、香料及合成药物中间体,全球每年生产的羰基化合物超过1 Mt,且大多由醇氧化而来[2]。传统的醇氧化方法是化学计量法,常用氧化剂为铬试剂(包括Jones试剂、重铬酸吡啶鎓、氯铬酸吡啶)[3-7]、锰试剂(氧化锰)[8-9]、钌的氧化物[10-11]、过钌酸四丙胺盐/N-甲基吗啉氧化物试剂[12-13]、活化的二甲基亚砜(DMSO)(Swern氧化)[14-15]、高价碘化物(Dess-Martin)[16-17]和Ag2CO3(Fetizon)[18]。但传统方法存在的缺点是:需要当量甚至过量的氧化剂,反应过程中易产生大量的重金属污染物;通常在含氯有机溶剂中进行,不仅后处理麻烦,且对环境污染很大;有些强氧化剂难以将反应产物控制在醛的阶段,往往使醛进一步深度氧化为酸。从环境保护和原子经济性的角度来看,上述方法必定会被改进或淘汰,因此开发绿色、高效、高选择性的醇氧化方法一直是研究热点。空气或O2是理想的氧化剂,它们不仅价格便宜,资源丰富,且氧化后的副产物为水。通常情况下O2处于稳定态,O2分子中两个未成对的单电子一旦被活化,其双自由基性质会促使形成高反应性及非选择性的自由基中间体,但中间体的活性往往较高,易发生深度氧化,从而导致产物的选择性降低。因此,需要开发温和条件下高选择性催化O2(空气)氧化醇体系。
O2与醇难以直接反应,需在催化剂作用下才能有效实现醇的选择性氧化,故催化剂的选择很关键。目前报道较多的是过渡金属催化剂和有机分子催化剂。有机分子催化剂主要有两种:一是稳定的自由基,常用2,2,6,6-四甲基哌啶氮氧自由基(TEMPO);二是N-羟基亚胺,常用N-羟基邻苯二甲酰亚胺生成的活泼自由基。氮氧自由基可与过渡金属或非金属催化剂组成有效的催化体系,能在温和条件下高选择性催化醇氧化。
本文主要综述了以分子氧为氧源的催化氧化醇体系,包括过渡金属(Cu,Fe,Co,V,Ru,Pd,Au)催化体系、非金属催化体系(TEMPO)及2,3-二氯-5,6-二氰对苯醌(DDQ)等其他体系。对各催化体系的适用范围和优缺点进行了分析,阐述了主要催化体系的催化机理,对已有研究成果进行了总结,并在此基础上对该领域的发展趋势作了展望。
1 过渡金属催化体系
1.1 Cu催化体系
早期Cu催化醇氧化反应主要使用的催化剂为单核铜酶(即半乳糖氧化酶)。半乳糖氧化酶可将D-半乳糖和许多伯醇催化氧化为相应的醛,同时将O2还原为H2O2(见式(1))[19]。半乳糖氧化酶的活性中心由一个单独的Cu(Ⅱ)离子、组氨酸咪唑基的氮、具有硫醚取代基的络氨酸自由基和一个络氨酸离子配位组成。半乳糖氧化酶催化氧化伯醇的反应机理见式(2)。
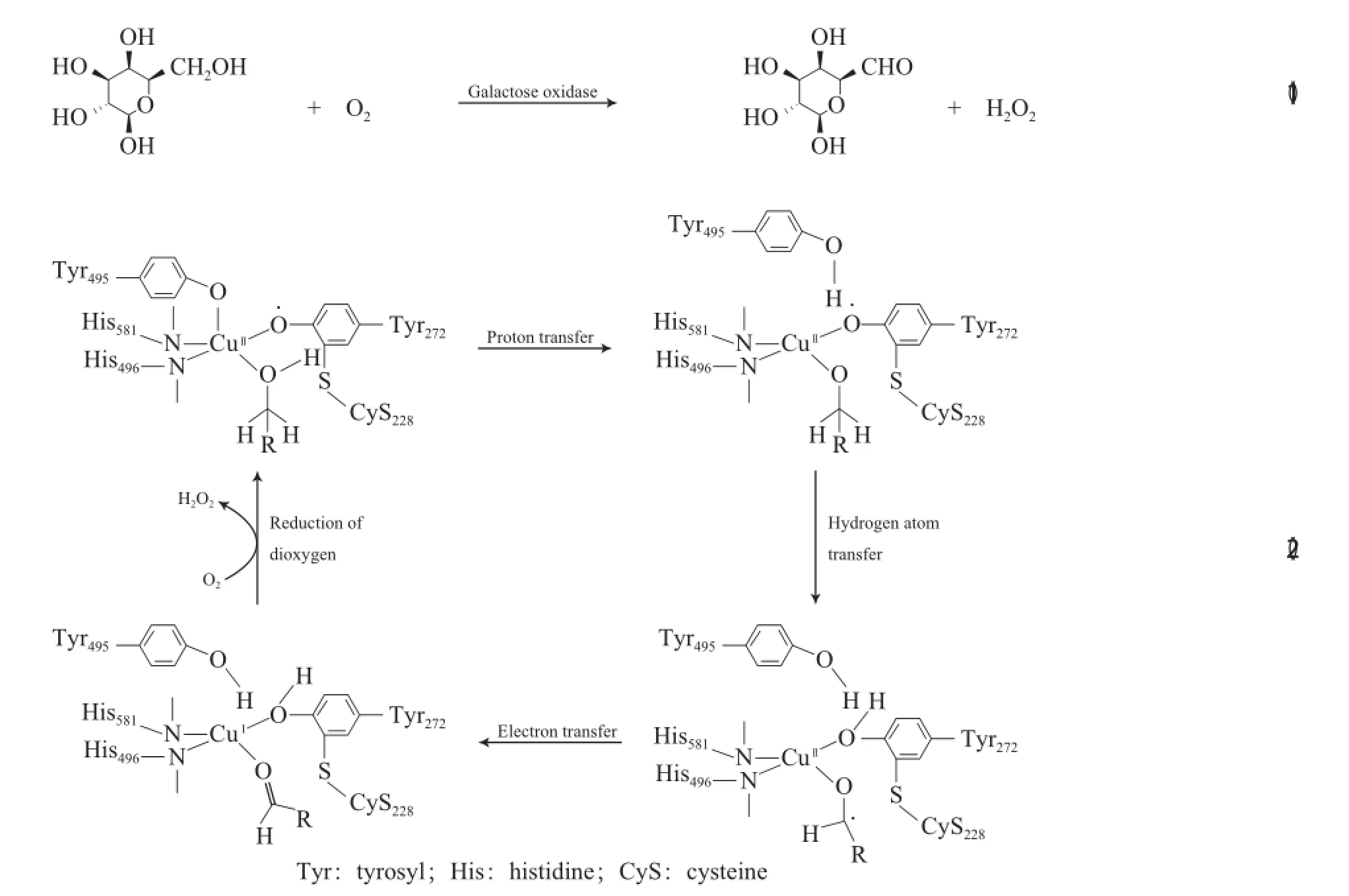
由于对催化反应起关键作用的是活性中心部分,因此用小分子铜配合物模拟该酶的反应性引起了许多研究者的兴趣,其中,最具代表性的模型化合物是由Stack等合成的[Cu(Ⅱ)BSP](BSP为联萘正四面配体)配合物[20],[Cu(Ⅱ)BSP]配合物能有效催化O2氧化苄基醇和烯丙醇(见式(3))。
Markó等[21]报道了Cu催化O2氧化醇体系,该体系包含5%(χ)CuCl、5%(χ)1,10-菲啰啉(Phen)、5%(χ)叠氮二羧酸二叔丁酯(DBADH2)、200%(χ)K2CO3、甲苯溶剂(记为CuCl/Phen/DBADH2体系),在空气或纯O2为氧化剂、70~90℃的条件下,CuCl/Phen/DBADH2体系能有效地氧化苄基和烯丙基醇为相应的醛或酮(见式(4)),对含杂原子(N和S)的醇同样具有很好的氧化效果,但该体系对脂肪醇尤其是脂肪伯醇的反应效果较差。随后他们用偶氮二甲酸二叔丁酯(DBAD)取代DBADH2[22]得到CuCl/Phen/ DBAD体系,在无氧条件下可有效地氧化各种醇(见式(5)),说明Cu-Phen在体系中起催化作用,当K2CO3用量从200%(χ)降至10%(χ)时,脂肪伯醇的选择性明显提高。使用O2为末端氧化剂时,碱的用量较大,为减少碱的用量,通过对无机碱的筛选发现仅K2CO3的效果最好。当CuCl/Phen/DBAD体系中的溶剂改成氟苯时(见式(6))[23],K2CO3的用量可降至25%(χ),该体系可将各种醇(如苄基伯醇、烯丙基伯醇、苄基仲醇、烯丙基仲醇、脂肪仲醇)有效地氧化为相应的醛或酮,但对脂肪伯醇的反应活性仍较低。随后Markó等在用氟苯为溶剂的CuCl/Phen/DBADH2体系中加入N-甲基咪唑(NMI),有效地实现了脂肪伯醇的氧化且未发现酸的生成(见式(7))[24]。对于用氟苯为溶剂的CuCl/Phen/DBAD体系,在无氧气和NMI时,脂肪伯醇同样可定量地转化为醛,但加入NMI后,反应反而被阻碍。

基于以上研究,Markó等提出了CuCl/Phen/ DBADH2体系催化醇氧化的反应机理(见图1)。从图1可看出,首先醇与3种化合物形成活性物种A,通过分子内的氢转移得到配合物B,接着释放产物醛并得到配合物C,配合物C迅速被O2捕获,生成物种D,物种D在加热下形成氢氧铜物种E,最后通过配体交换和消除水再形成活性物种A。其中,配合物C上有空配位点,尽管配合物C通常能迅速与O2反应,但偶尔会有醇发生竞争配位,形成物种F,物种F经H转移得到非活性的配合物G。因此,加入NMI后,α位存在空间位阻的醇能被高效地氧化为相应的醛和酮,而对α位无取代基的脂肪伯醇的反应效果较差。
Semmelhack等[25]报道了CuCl/TEMPO(10%(χ)CuCl+10%(χ)TEMPO)体系,该体系在溶剂为二甲基甲酰胺(DMF)、O2鼓泡、室温的条件下,能有效地氧化活泼伯醇(见式(8)),但对仲醇的氧化速率明显比伯醇慢。Semmelhack等提出的CuCl/TEMPO 体系催化醇氧化的反应机理见图2。从图2可看出,Cu(Ⅱ)通过单电子转移将TEMPO氧化为氮氧正离子TEMPO+; TEMPO+是氧化醇的真正氧化剂,并生成对应的羰基化合物和TEMPOH;TEMPOH和TEMPO+再迅速反应生成TEMPO;Cu(Ⅰ)在H+的存在下被O2氧化为Cu(Ⅱ),并有水生成,生成的Cu(Ⅱ)重新进入循环。
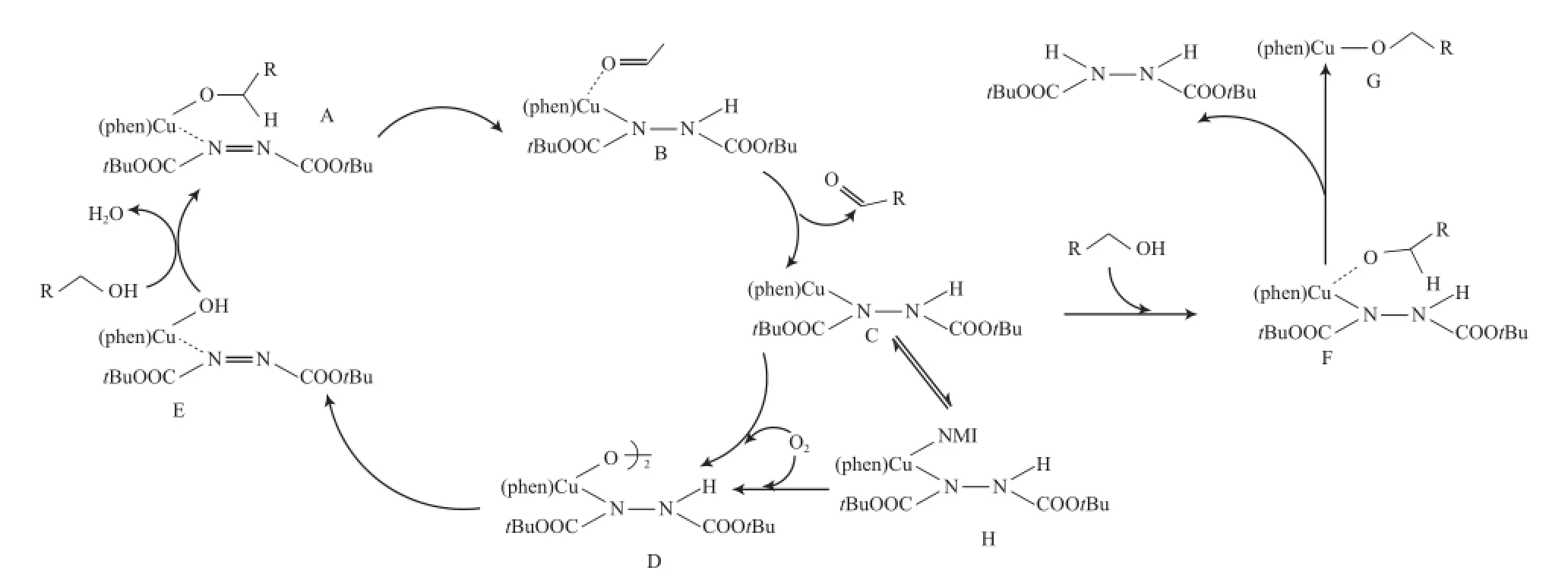
图1 Markó等提出的CuCl/Phen/DBADH2体系催化醇氧化的机理Fig.1 Mechanism for the aerobic alcohol oxidation catalyzed by CuCl/Phen/DBADH2.

图2 Semmelhack等提出的CuCl/TEMPO体系催化醇氧化的反应机理[25]Fig.2 Mechanism for the aerobic alcohol oxidation catalyzed by CuCl/TEMPO proposed by Semmelhack[25].
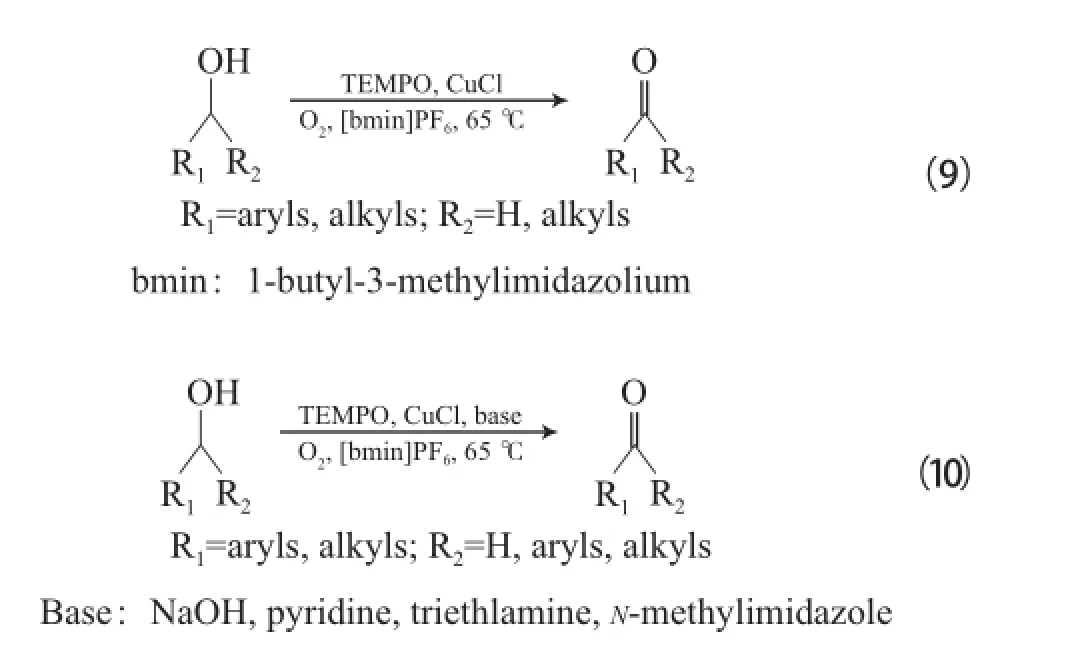
Ansari等[26]采用CuCl/TEMPO体系,以离子液体为溶剂将伯仲醇氧化为相应的醛或酮(见式(9)),离子液体可循环使用,但反应时间较长。
Liu等[27]在以离子液体为溶剂的CuCl/TEMPO体系中加入碱后发现,反应速率明显得到提高(反应见式(10))。
Dijksman等[28]对CuCl/TEMPO体系催化氧化醇的反应机理研究后认为,反应过程中未发现氮氧正离子TEMPO+的生成。这是因为,TEMPO+作为氧化剂能有效氧化各种醇 ,而该体系对简单的脂肪醇反应活性较低,而且在该体系的反应条件下,TEMPOH能迅速被O2氧化为TEMPO。因此,他们提出了另外一种反应机理,反应机理见图3。从图3可看出,反应机理是以Cu为中心的醇氧化过程:通过单电子转移,Cu(Ⅰ)被TEMPO氧化为Cu(Ⅱ)-TEMPO物种;接着醇取代TEMPO形成Cu醇盐和TEMPOH;另一分子的TEMPO与Cu醇盐配位,通过分子内β-H消除得到产物和TEMPOH;TEMPOH再被O2迅速氧化为TEMPO。
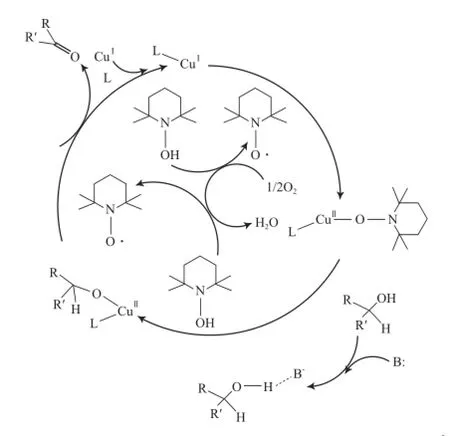
图3 Sheldon等提出的CuCl/TEMPO体系催化醇氧化的反应机理[28]Fig.3 Sheldon’s mechanism for the aerobic alcohol oxidation catalyzed by CuCl/TEMPO[28].
Gamez等[29]报道了CuBr2(2,2-bpy)/TEMPO/ t-BuOK(bpy为联吡啶)体系,该体系以乙腈/水为溶剂,室温下可有效氧化伯醇为相应的醛(见式(11),但对活泼和非 活泼的仲醇均无催化效果。反应机理见图4。
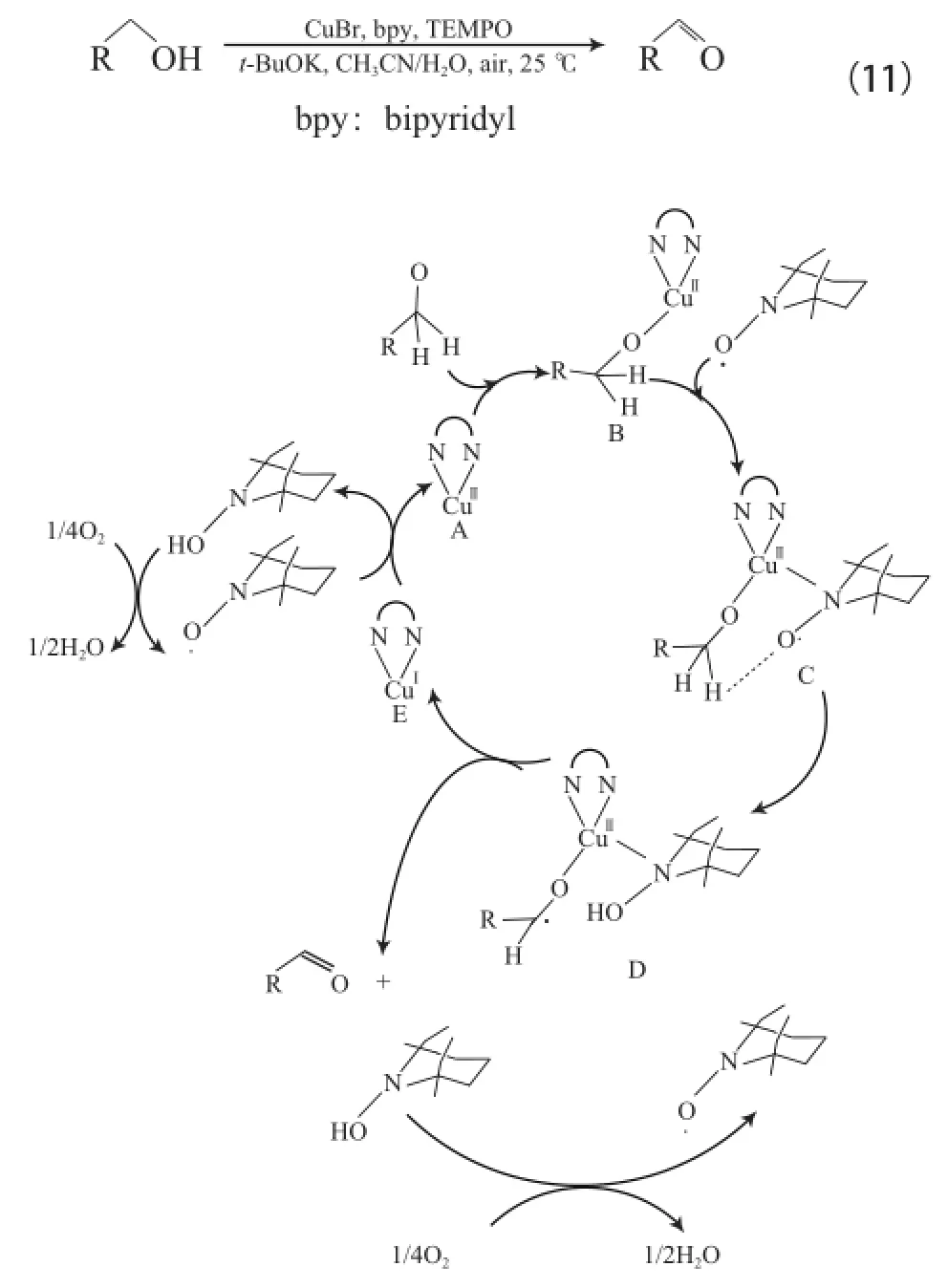
图4 CuBr2(2,2-bpy)/TEMPO/t-BuOK催化伯醇的氧化机理Fig.4 Proposed mechanism for the oxidation of primary alcohols catalyzed by CuBr2(2,2-bpy)/TEMPO/t-BuOK.
从图4可看出,TEMPO可能为H的受体。醇盐与Cu(Ⅱ)配合物A形成烷氧基物种B;TEMPO很有可能与Cu(Ⅱ)离子以η2的方式螯合形成物种C,然后通过β-H转移到TEMPO得到自由基物种D;再通过分子内单电子转移得到产物醛、TEMPOH和Cu(Ⅰ)物种E;通过TEMPO的调节,物种E再被氧化为配合物A,TEMPOH则被O2氧化为TEMPO,完成催化循环。仲醇不能被氧化可能是因为甲基空间位阻的存在不利于物种C的形成,伯醇能有效地被氧化还可能是因为另一个β-H稳定了自由基物种D。
Jiang等[30]报道了Cu(ClO4)2/4-二甲氨基吡啶(DMAP)/acetamido-TEMPO三组分体系,在以离子液体[bmpy]PF6为溶剂、室温、0.1 MPa O2的条件下,可有效地氧化活泼伯醇为醛(见式(12)),且循环5次使用后,反应活性无明显降低。该体系的反应机理与CuBr2(2,2-bpy)/TEMPO/t-BuOK体系类似,由于存在空间位阻,该体系对仲醇的反应活性低。

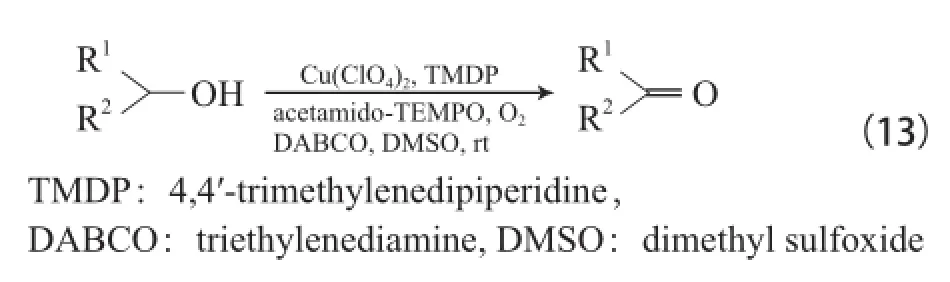
Jiang等[31]还报道了Cu(ClO4)2/4,4-丙基哌啶基哌啶/乙酰胺基-TEMPO/三乙烯二胺(DABCO)四组分体系,在DMSO为溶剂和室温下能有效氧化各种醇为相应的醛或酮(见式(13)),由于使用强极性DMSO溶剂,该体系可循环使用3次且反应活性无明显降低。DBACO在该体系中可能有两个作用:一是作为碱夺取醇羟基上的质子;二是作为弱的含N配体与Cu2+离子配位。该体系与Cu(ClO4)2/DMAP/乙酰胺基-TEMPO和CuBr2(2,2-bpy)/TEMPO/ t-BuOK体系相比,由于加入了DABCO,延长反应时间能有效地氧化仲醇为相应的酮。
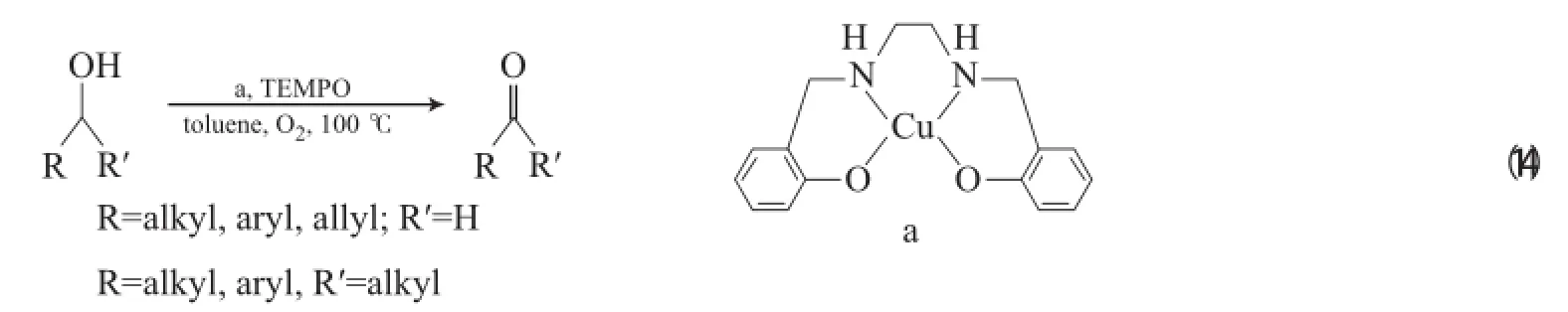
Velusamy等[32]报道了Cu-salen/TEMPO体系,在以甲苯为溶剂、0.1 MPa O2和100℃的条件下,有效地氧化伯醇为相应的醛(见式(14))。由于采用非极性甲苯为溶剂,该体系循环3次后反应活性无明显降低。其反应机理与CuBr2(2,2-bpy)/TEMPO/t-BuOK体系一样,对仲醇无反应活性,也可能是因为存在空间位阻。
Kumpulainen等[33]研究Cu-TEMPO体系时发现,加入有机碱1,8-二氮杂二环十一碳-7-烯(DBU)和NMI能显著提高催化效率,乙腈为溶剂时反应效果最好(见式(15))。Cu-TEMPO-DBU-NMI体系对TEMPO是一级反应,对Cu盐是二级反应,能高效氧化脂肪伯醇,但对仲醇的氧化性能较低。

Hoover等[34]研究发现,Cu-TEMPO体系在以NMI为碱、乙腈为溶剂时,Cu+盐的催化效果明显优于Cu2+盐,在室温和空气氛围下,能高效高选择性氧化各种伯醇(包括烯丙基、苄基、脂肪醇)为相应的醛(见式(16)),还能氧化带其他官能团的醇,特别对二元醇,不需保护即能高选择性氧化伯醇。

Hoover等[35-36]对(bpy)CuI(OTf)/TEMPO体系的反应机理进行了研究,反应机理见图5。从图5可看出,活泼醇和非活泼醇具有不同的决速步骤。活泼醇氧化时,催化剂的氧化过程是决速步骤,催化剂氧化经过双核Cu2O2中间体;非活泼伯醇氧化时,底物氧化和催化剂氧化影响反应速率。由于脂肪醇的解离常数较大,不容易形成Cu-烷氧化合 物,而且具有强α C—H键,均明显影响产物的形成。O2首先与一个Cu+反应生成Cu2+超氧化物,接着在另一个Cu+中心上形成过氧化物桥联的双核铜Cu2O2,Cu2O2被认为能够氧化TEMPOH为TEMPO,通过H转移形成CuⅡ—OOH物种和CuⅠ副产物。接着,CuⅡ—OOH与H2O反应得到CuⅡ—OH并释放H2O2,在一定的反应条件下,H2O2迅速分解为H2O和O2。通过底物与CuⅡ形成Cu-烷氧化合物和TEMPO夺H实现底物氧化。

图5 CuI/TEMPO催化醇氧化循环过程Fig.5 Catalytic cycle for the aerobic oxidation of alcohols catalyzed by CuI/TEMPO.
Cu-TEMPO体系对仲醇氧化的效果较差,这是因为:仲醇本身空间位阻大,而且在最后一步Cu-烷氧化合物和TEMPO共同与底物作用时,TEMPO的空间位阻较大。为实现仲醇氧化,Steves等[37]将TEMPO换成空间位阻小的氮氧自由基9-氮杂双环[3.3.1]壬烷-N-氧自由基(ABNO),催化体系5%(χ)Cu(MeCN)4OTf、5%(χ)4,4’-DMeO-2,2’-bpy(DMeO为二甲氧基)、10%(χ)NMI和1%(χ)ABNO组成,可有效氧化各种类型的仲醇,对伯醇也有非常好的氧化效果(见式(17))。

Cu-TEMPO体系多使用联吡啶类配体,Zhang等[38]研究发现,以L-脯氨酸为配体时,Cu-TEMPO体系也能实现活泼伯仲醇的氧化。催化体系由5%(χ)CuI、5%(χ)L-脯氨酸、5%(χ)TEMPO、100%(χ)tBuOK组成,在DMF为溶剂、室温和空气氛围的条件下,能将仲醇氧化为相应的酮。催化体系由5%(χ)CuBr、5%(χ)L-脯氨酸、5%(χ)TEMPO和100%(χ)Na2CO3组成,在甲醇为溶剂、室温和空气氛围的条件下,能将伯醇氧化为相应的醛。
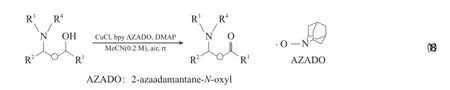
Sasano等[39]采用Cu/2-氮杂金刚烷-N-氧自由基(AZADO)体系有效实现了非保护胺基醇的氧化(见式(18)),进一步证明了空间位阻小的氮氧自由基氧化效果优于TEMPO。当AZADO的用量(χ)为1%~5%时,即可有效氧化伯仲胺基醇。与传统体系相比,Cu/AZADO催化体系的效果更好,醇的O—H酸性比胺的N—H酸性强,有利于与Cu形成Cu-烷氧物种,进一步实现H原子转移到氮氧自由基上。
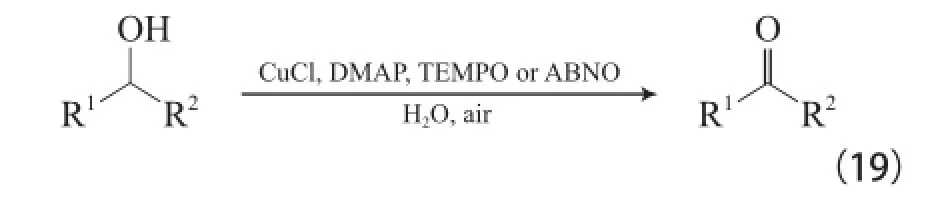
Cu-TEMPO体系多在乙腈或其他有机溶剂中进行,Zhang等[40]研究发现,当溶剂为水时,CuCl/DMAP/TEMPO(或ABNO)体系能在空气氛围和室温条件下实现各种活泼伯仲醇的氧化(见式(19))。
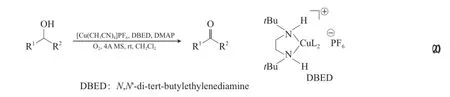
Cu-TEMPO体系的催化活性非常高,尤其对于非活泼醇的氧化,但TEMPO或其他氮氧自由基(如ABNO和AZADO)的价格昂贵、制备复杂。Xu等[41]模拟酪氨酸酶开发了无TEMPO的Cu-N,N-双(叔丁基)乙烯二胺(DBED)/DMAP体系,该体系能在室温下实现非活泼脂肪伯醇高选择性的氧化为醛(见式(20))。两种体系的底物选择性不同,当伯仲醇同时存在时,Cu-DBED体系优先氧化活泼仲醇,而Cu-TEMPO体系优先氧化伯醇,说明Cu-DBED体系受底物空间位阻的影响小。
1.2 Fe催化体系
尽管Fe具有很好的氧化还原性,但Fe参与的醇氧化体系较少,主要是Fe/TEMPO体系。不同于Cu/TEMPO体系,Fe/TEMPO体系通常不需加入任何额外的碱或有机配体,即使在1,2-二氯乙烷溶剂中,也能取得很好的催化效果,而Cu/TEMPO体系通常在乙腈溶剂中进行反应。Fe/TEMPO体系可有效催化各种醇,包括含双键、三键及杂原子的醇。对于Fe/TEMPO体系催化醇氧化机理的研究很少。铁盐催化剂主要是Fe(NO3)3(或添加NaNO2),催化过程中产生的NOx起重要的作用:NO2能氧化TEMPO为TEMPO+,因此体系中需加入硝酸盐为添加剂;NO2也能协助Fe2+氧化为Fe3+。Fe/TEMPO体系还能顺利氧化仲醇,这是因为:该体系生成了TEMPO+氧化剂,且体系中不加额外配体,相对Cu/TEMPO体系的空间位阻小。
Martin等[42]首先报道了Fe催化的醇氧化体系,该体系采 用Fe(NO3)3和FeBr3为催化剂、在乙腈为溶剂、空气氛围和室温条件下能有效地氧化脂肪仲醇和苄基伯醇为相应的酮或醛 (见式(21))。该体系对脂肪伯醇无反应活性,但可选择性氧化仲醇。

Wang等[43]报道了FeCl3・6H2O/TEMPO/NaNO2体系。该体系在室温和空气氛围下可氧化各种醇为相应的醛或酮,产物选择性大于99%(见式(22)),在硫醚存在下能选择性氧化醇,反应机理见图6。从图6可看出,FeⅢ-TEMPO为氧化醇的活性物种,可转化为FeII-TEMPOH,NaNO2分解的NO2再将FeII-TEMPOH氧化为FeⅢ-TEMPO并还原为NO,NO又能迅速被O2氧化为NO2,从而完成催化循环。
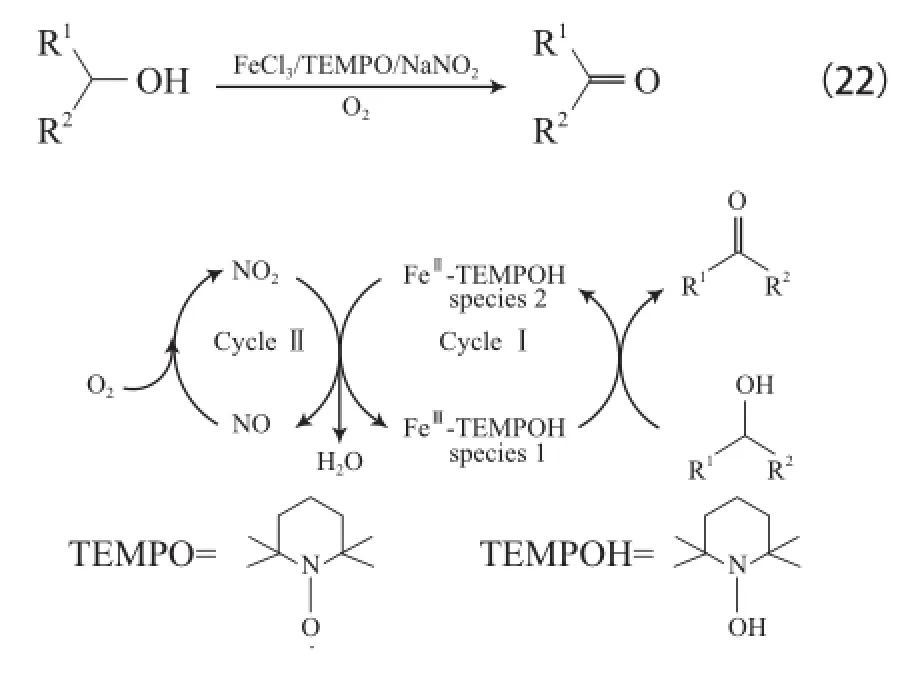
图6 FeCl3・6H2O/TEMPO/NaNO2体系催化醇氧化的机理Fig.6 Proposed mechanism for the aerobic alcohol oxidation catalyzed by FeCl3・6H2O/TEMPO/NaNO2.
Wang等[44]还报道了Fe(NO3)3/4-OH-TEMPO体系,即用价格便宜的4-OH-TEMPO取代了价格昂贵的TEMPO。该体系在乙腈为溶剂、室温、空气条件下可氧化各种醇为相应的醛或酮(见式(23))。该体系的反应机理与FeCl3・6H2O/ TEMPO/NaNO2体系相似,是NO2的来源。

由于FeCl3・6H2O/TEMPO/NaNO2体系对脂肪伯醇的氧化效果较差,Yin等[45]对体系进行改进得到FeCl3/4-acetamido-TEMPO/NaNO2体系,4-acetamido-TEMPO的用量可降至0.1%(x),该体系可氧化各种伯醇(包括脂肪伯醇)为相应的醛(见式(24))。

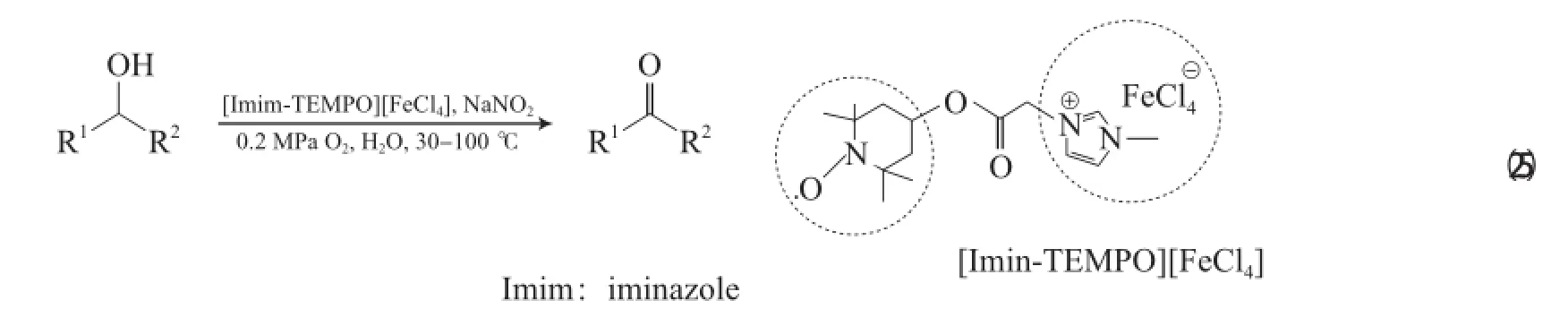
Miao等[46]研究发现,具有磁性的离子液体[Imim-TEMPO][FeCl4](Imim为咪唑盐)加入5%(χ)NaNO2和H2O,在0.2 MPa O2和 30~100℃的条件下,可将苄基醇氧化为相应的醛或酮(见式(25)),该体系对含杂原子(N,O,S)的芳香醇同样具有很好的氧化效果。但该体系不能氧化脂肪醇,故该体系的反应机理不同于FeCl3・6H2O/ TEMPO/NaNO2体系。EPR表征结果显示,该体系在反应过程中原位产生了自由基物种。ESI-MS表征结果显示,反应过程中Fe的价态发生了变化,经历了由[Imim-TEMPO][FeCl4]和NaNO2电子传递的过程。
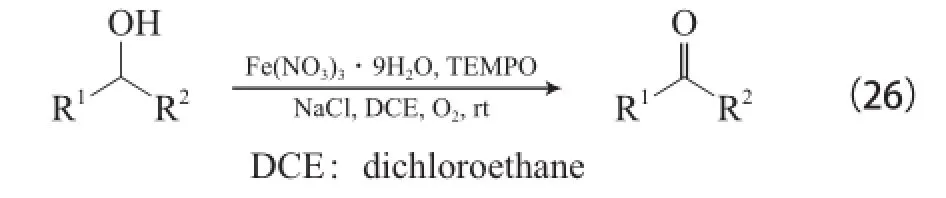
Ma等[47-49]报道了Fe(NO3)3・9H2O/TEMPO/ NaCl体系,该体系以二氯乙烷为溶剂,在O2和室温的条件下能氧化脂肪伯仲醇、苄基伯仲醇和烯丙基醇为相应的醛或酮(见式(26)),该体系还可用于炔丙基醇和联烯醇的氧化,反应效果较好。
本课题组[50]首次报道了不加Fe(NO3)3或NaNO2,而加入SiO2的FeCl3・6H2O/TEMPO体系。该体系能催化O2(或空气)将醇选择性氧化为相应的醛或酮(见式27)。研究结果表明,SiO2不但可加快反应速率,还可提高产物选择性。这可能是因为,底物和催化剂吸附在Si—OH表面形成了特殊反应区域。采用Uv-vis光谱法研究了该体系的反应过程,表征结果显示,FeⅢ-TEMPO配合物可能是反应的活性中间体。催化体系由8%(χ)FeCl3・6H2O、2%~5%(χ)TEMPO、0.2 g SiO2组成,在0.5 MPa O2和80℃的条件下,产物的最高收率为99%。在优化的条件下,含双键的肉桂醇的反应转化率大于99%,肉桂醛收率80%。对于脂肪仲醇,增加FeCl3・6H2O和TEMPO的用量,产物酮的收率可达67%~90%。利用空气代替纯O2时,在相同的催化剂用量和反应温度下,延长反应时间也可得到很好的反应效果,产物的最高收率能达到95%。

但该体系不能氧化脂肪伯醇。随后,本课题组[51]通过化学方法用烷基化试剂对SBA-15分子筛进行修饰,然后通过化学反应把TEMPO键合到SBA-15分子筛上得到SBA-15-TEMPO。将SBA-15-TEMPO结合FeCl3・6H2O及助催化剂NaNO2组成了三组分体系用于催化O2氧化醇反应。当SBA-15-TEMPO用量(χ)为0.1%~1%时,能有效地氧化脂肪仲醇和芳香伯仲醇为相应的醛和酮。含N杂原子的醇也能被氧化为相应的醛。但对脂肪伯醇和环己醇的催化效果较差。如采用空气代替纯O2时,在相同的催化剂用量和反应温度下,延长反应时间也可得到很好的反应效果。氧化苯甲醇时,SBA-15-TEMPO用量降至0.01%(χ)时反应仍可完成,转化率大于99%,苯甲醛收率为97.7%。SBA-15-TEMPO可循环使用13次,反应活性略降低。由于FeCl3・6H2O在甲苯中不溶解,SBA-15-TEMPO和FeCl3・6H2O两组分同时循环使用,循环4次,反应活性无明显降低。
[1]Hudlicky M.Oxidations in Organic Chemistry[M].Washington DC:American Chemical Society,1990:35-76.
[2]Zhan Bizeng,Thompson A.Recent Developments in the Aerobic Oxidation of Alcohols[J].Tetrahedron,2004,60(13):2917-2935.
[3]Holum J R.Study of the Chromium(Ⅵ) Oxide-Pyridine Complex[J].J Org Chem,1961,26:4814-4816.
[4]Lee D G,Spitzer U A.Aqueous Dichromate Oxidation of Primary Alcohols[J].J Org Chem,1970,35(10):3589-3590.
[5]Cainelli G,Cardillo G.Chromium Oxidants in Organic Chemistry[M].Berlin:Springer,1984.
[6]Ley S V,Madin A.Comprehensive Organic Synthesis[M]// Trost B M,Fleming I,Ley S V,Eds.Pergamon:Oxford,1991,7:251-289.
[7]Corey E J,Suggs J W.Pyridinium Chlorochromate:Efficient Reagent for Oxidation of Primary and Secondary Alcohols to Carbonyl Compounds[J].Tetrahedron Lett,1975(31):2647-2650.
[8]Regen S L,Koteel C.Activation Through Impregnation:Permanganate-Coated Solid Supports[J].J Am Chem Soc,1977,99(11):3837-3838.
[9]Menger F M,Lee C.Synthetically Useful Oxidations at Solid Sodium Permanganate Surfaces[J].Tetrahedron Lett,1981,22(18):1655-1656.
[10]Berkowitz L M,Rylander P N.Use of Ruthenium Tetroxide as a Multi-Purpose Oxidant[J].J Am Chem Soc,1958,80:6682-6684.
[11]Griffith W P.Ruthenium Oxo Complexes as Organic Oxidants[J].Chem Soc Rev,1992,21(3):179-185.
[12]Griffith W P,Ley S V,Whitcombe G P,et al.Preparation and Use of Tetrabutylammonium Perruthenate (TBAP Reagent) and Tetrapropylammonium Perruthenate (TPAP Reagent) as New Catalytic Oxidants for Alcohols[J].J Chem Soc,Chem Commun,1987(21):1625-1627.
[13]Ley S V,Norman J,Griffith W P,et al.Tetrapropylammonium Perruthenate,Pr4N+RuO4-TPAP:A Catalytic Oxidant for Organic Synthesis[J].Synthesis,1994(7):639-666.
[14]Albright J D,Goldman L.Dimethyl Sulfoxide-Acid Anhydride Mixtures.New Reagents for Oxidation of Alcohols[J].J Am Chem Soc,1965,87(18):4214-4216.
[15]Pfitzner K E,Moffatt J G.Sulfoxide-Carbodiimide Reactions:Ⅰ.A Facile Oxidation of Alcohols[J].J Am Chem Soc,1965,87(24):5661-5670.
[16]Dess D B,Martin J C.Readily Accessible 12-I-5 Oxidant for the Conversion of Primary and Secondary Alcohols to Aldehydes and Ketones[J].J Org Chem,1983,48(22):4155-4156.
[17]Dohi T.Development of Environmentally Benign Oxidations Using Hypervalent Iodine (Ⅲ) Reagents[J].Yakugaku zasshi:J Pharm Soc Jpn,2006,126(9):757-766.
[18]Li Tsung Tee,Wu Yulin,Walsgrove T C.A Facile Total Synthesis of Racemic Aklavinonet[J].Tetrahedron,1984,40(22):4701-4710.
[19]Whittaker J W.Metal Ions in Biological Systems[M]//Sigel H,Sigel A,Eds.New York:Marcel Dekker,1994:315-360.
[20]Wang Yadong,DuBois J L,Hedman B,et al.Catalytic Galactose Oxidase Models:Biomimetic Cu(Ⅱ)-Phenoxyl-Radical Reactivity[J].Science,1998,279(5350):537-540.
[21]Markó I E,Giles P R,Tsukazaki M,et al.Copper-Catalyzed Oxidation of Alcohols to Aldehydes and Ketones:An Efficient Aerobic Alternative[J].Science,1996,274(5295):2044-2046.
[22]Markó I E,Tsukazaki M,Giles P R,et al.Anaerobic Copper-Catalyzed Oxidation of Alcohols to Aldehydes and Ketones[J].Angew Chem Int Ed,1997,36(20):2208-2210.
[23]Markó I E,Gautier A,Chelle-Regnaut I,et al.Efficient and Practical Catalytic Oxidation of Alcohols Using Molecular Oxygen[J].J Org Chem,1998,63(22):7576-7577.
[24]Markó I E,Gautier A,Dumeunier R,et al.Efficient,Copper-Catalyzed,Aerobic Oxidation of Primary Alcohols[J].Angew Chem Int Ed,2004,43(12):1588-1591.
[25]Semmelhack M F,Schmid C R,Cortes D A,et al.Oxidation of Alcohols to Aldehydes with Oxygen and Cupric Ion,Mediated by Nitrosonium Ion[J].J Am Chem Soc,1984,106(11):3374-3376.
[26]Ansari I A,Gree R.TEMPO-Catalyzed Aerobic Oxidation of Alcohols to Aldehydes and Ketones in Ionic Liquid[bmim][PF6][J].Org Lett,2002,4(9):1507-1509.
[27]Liu Lin,Ji Liuyan,Wei Yunyang.Based-Promoted Aerobic Oxidation of Alcohols to Corresponding Aldehydes or Ketones Catalyzed by CuCl/TEMPO[J].Catal Commun,2008,9(6):1379-1382.
[28]Dijksman A,Arends I W C E,Sheldon R A.Cu(Ⅱ)-Nitroxyl Radicals as Catalytic Galactose Oxidase Mimics[J].Org Biomol Chem,2003,1(18):3232-3237.
[29]Gamez P,Arends I W C E,Reedijk J,et al.Copper(Ⅱ)-Catalysed Aerobic Oxidation of Primary Alcohols to Aldehydes[J].Chem Commun,2003(19):2414-2415.
[30]Jiang Nan,Ragauskas A J.Copper (Ⅱ)-Catalyzed Aerobic Oxidation of Primary Alcohols to Aldehydes in Ionic Liquid[bmpy]PF6[J].Org Lett,2005,7(17):3689-3692.
[31]Jiang Nan,Ragauskas A J.Cu (Ⅱ)-Catalyzed Selective Aerobic Oxidation of Alcohols Under Mild Conditions[J].J Org Chem,2006,71(18):7087-7090.
[32]Velusamy S,Srinivasan A,Punniyamurthy T.Copper (Ⅱ)Catalyzed Selective Oxidation of Primary Alcohols to Aldehydes with Atmospheric Oxygen[J].Tetrahedron Lett,2006,47(6):923-926.
[33]Kumpulainen E T T,Koskinen A M P.Catalytic Activity Dependency on Catalyst Components in Aerobic Copper-TEMPO Oxidation[J].Chem Eur J,2009,15(41):10901-10911.
[34]Hoover J M,Stahl S S.Highly Practical Copper (Ⅰ)/TEMPO Catalyst System for Chemoselective Aerobic Oxidation of Primary Alcohols[J].J Am Chem Soc,2011,133(42):16901-16910.
[35]Hoover J M,Ryland B L,Stahl S S.Mechanism of Copper(Ⅰ)/TEMPO-Catalyzed Aerobic Alcohol Oxidation[J].J Am Chem Soc,2013,135(6):2357-2367.
[36]Hoover J M,Ryland B L,Stahl S S.Copper/TEMPO-Catalyzed Aerobic Alcohol Oxidation:Mechanistic Assessment of Different Catalyst Systems[J].ACS Catal,2013,3(11):2599-2605.
[37]Steves J E,Stahl S S.Copper(Ⅰ)/ABNO-Catalyzed Aerobic Alcohol Oxidation:Alleviating Steric and Electronic Constraints of Cu/TEMPO Catalyst Systems[J].J Am Chem Soc,2013,135(42):15742-15745.
[38]Zhang Guofu,Han Xingwang,Luan Yuxin,et al.L-Proline:An Efficient N,O-Bidentate Ligand for Copper-Catalyzed Aerobic Oxidation of Primary and Secondary Benzylic Alcohols at Room Temperature[J].Chem Commun,2013,49(72):7908-7910
[39]Sasano Y,Nagasawa S,Yamazaki M,et al.Highly Chemoselective Aerobic Oxidation of Amino Alcohols into Amino Carbonyl Compounds[J].Angew Chem Int Ed,2014,53(12):3236-3240.
[40]Zhang Guoqi,Yang Chengxiong,Liu E,et al.Mild,Green Copper/4-Dimethylaminopyridine Catalysed Aerobic Oxidation of Alcohols Mediated by Nitroxyl Radicals in Water[J].RSC Adv,2014,4(106):61907-61911.
[41]Xu Boran,Lumb J P,Arndtsen B A.A TEMPO-Free Copper-Catalyzed Aerobic Oxidation of Alcohols[J].Angew Chem Int Ed,2015,56(14):4208-4211.
[42]Martin S E,Suarez D E.Catalytic aerobic oxidation of Alcohols by Fe(NO3)3-FeBr3[J].Tetrahedron Lett,2002,43(25):4475-4479.
[43]Wang Naiwei,Liu Renhua,Chen Jiping,et al.NaNO2-Activated,Iron-TEMPO Catalyst System for Aerobic Alcohol Oxidation Under Mild Conditions[J].Chem Commun,2005(42):5322-5324.
[44]Wang Xinliang,Liang Xinmiao.Aerobic Oxidation of Alcohols to Carbonyl Compounds Catalyzed by Fe(NO3)3/4-OH-TEMPO Under Mild Conditions[J].催化学报,2008,29(9):935-939.
[45]Yin Weili,Chu Changhu,Lu Qiongqiong,et al.Iron Chloride/4-Acetamido-TEMPO/Sodium Nitrite-Catalyzed Aerobic Oxidation of Primary Alcohols to the Aldehydes[J].Adv Synth Catal,2010,352(1):113-118.
[46]Miao Chenxia,Wang Jinquan,Yu Bing,et al.Synthesis of Bimagnetic Ionic Liquid and Application for Selective Aerobic Oxidation of Aromatic Alcohols Under Mild Conditions[J].Chem Commun,2011,47(9):2697-2699.
[47]Ma Shengming,Liu Jinxian,Li Suhua,et al.Development of a General and Practical Iron Nitrate/TEMPO Catalyzed Aerobic Oxidation of Alcohols to Aldehydes/Ketones:Catalysis with Table Salt[J].Adv Synth Catal,2011,353(6):1005-1017.
[48]Liu Jinxian,Ma Shengming.Iron-Catalyzed Aerobic Oxidation of Allylic Alcohols:The Issue of C-C Bond Isomerization[J].Org Lett,2013,15(20):5150-5153.
[49]Liu Jinxian,Ma Shengming.Aerobic Oxidation of Indole Carbinols Using Fe(NO3)3・9H2O/TEMPO/NaCl as Catalysts[J].Org Biomol Chem,2013,11(25):4186-4193.
[50]Wang Lianyue,Li Jun,Lü Ying,et al.Selective Aerobic Oxidation of Alcohols Catalyzed by Iron Chloride Hexahydrate/ TEMPO in the Presence of Silica Gel[J].Appl Organomet Chem,2012,26(1):37-43.
[51]Wang Lianyue,Li Jun,Zhao Xiaoping,et al.An Efficient and Scalable Room Temperature Aerobic Alcohol Oxidation Catalyzed by Iron Chloride Hexahydrate/Mesoporous Silica Supported TEMPO[J].Tetrahedron,2013,69(30):6041-6045.
(待续)
(编辑 邓晓音)
Advances in Homogeneous Catalytic Oxidation of Alcohols by Molecular Oxygen
Wang Lianyue,Gao Shuang
(Dalian National Laboratory for Clean Energy,Dalian Institute of Chinese Physics,Chinese Academy of Science,Dalian Liaoning 116023,China)
Advances in the selective alcohol oxidation with molecular oxygen catalyzed by homogeneous catalyst systems,namely homogeneous transition metal(Cu,Fe,Co,V,Ru,Pd,Au) catalytic systems,non-metal catalytic system(2,2,6,6-tetramethyl-piperidyl-1-oxy) and small organic molecule catalytic systems(2,3-dichloro-5,6-dicyano-1,4-benzoquinone),to aldehydes or ketones were reviewed.The application,advantages and disadvantages of the catalytic systems were discussed.The possible catalytic mechanisms of the main catalytic systems were focused on.Based on the existing researches,the development trends in this field were also forecasted.
molecular oxygen;alcohol;selective oxidation;homogeneous catalysis
1000-8144(2015)11-1277-11
TQ 203.5
A
2015-03-30;[修改稿日期]2015-08-23。
王连月(1984—),男,山东省聊城市人,博士,助理研究员,电话 0411-84379726,电邮 lianyuewang@dicp.ac.cn。联系人:高爽,电话 0411-84379248,电邮 sgao@dicp.ac.cn。
国家自然科学基金资助项目(21403219)。
