超高效液相色谱法测定卡托普利片中卡托普利二硫化物含量
2015-01-05谭金峰马云云
谭金峰,马云云
(1.潍坊市环境监测中心站,山东潍坊 261041; 2潍坊市食品药品检验检测中心,山东潍坊 261041)
超高效液相色谱法测定卡托普利片中卡托普利二硫化物含量
谭金峰1,马云云2
(1.潍坊市环境监测中心站,山东潍坊 261041; 2潍坊市食品药品检验检测中心,山东潍坊 261041)
采用超高效液相色谱法测定卡托普利片中卡托普利二硫化物的含量。用Waters AcquityUPLC BEH C18色谱柱,以乙腈-水(pH 3.0)为流动相,流速0.2 mL/min,进样量为5 μL,外标法以峰面积定量。卡托普利二硫化物的质量浓度在0.2~10 μg/mL范围内与色谱峰面积呈良好的线性关系,r>0.999 95,峰面积测定结果的相对标准偏差为0.65% (n=6),加标回收率为98.9%,二硫化物与主峰的分离度大于6.0。用该法对4批样品进行检测,卡托普利二硫化物含量为0.3%~3.8%。与常规液相色谱法比较,该法分离效率高,重复性好,可用于卡托普利片的质量控制。
超高效液相色谱法;卡托普利二硫化物;含量测定;质量控制;快速检验
卡托普利片为抗高血压药物,是血管紧张素转化酶抑制剂,临床上被广泛应用,属于国家基本药物目录品种。制剂中卡托普利二硫化物(以下简称二硫化物)是一种已知杂质,目前行业内普遍采用《中国药典》规定的高效液相色谱法测定,外标法定量。按照《中国药典》中的色谱条件[1]测定二硫化物出峰慢、峰形差、理论板数低、使用大粒径填料、耗时、难以快速定量,即使采用梯度洗脱也需运行35 min[2],还会导致基线波动,影响准确定量,而且梯度洗脱较等度洗脱重现性差。目前中国药典中药品快速检验技术[3]不断增加,在测定卡托普利片中卡托普利二硫化物时需要有效手段快速分离二硫化物并准确定量。超高效液相色谱法在药物分析、食品检测、环境监测等方面的研究已有不少报道[4-15],但目前还未见采用超高效液相色谱法测定卡通托普利中二硫化物的报道。笔者采用该法建立了二硫化物的快速分析方法,方法操作简便,结果准确,分离度与重复性好,分析效率高,可为卡托普利质量控制提供参考方法。
1 实验部分
1.1 主要仪器与试剂
液相色谱仪:Waters AcquityTM UPLC型,配有高压泵,双波长紫外检测器,自动进样器,柱温箱,Empower Pro色谱工作站,美国Waters公司;
电子天平:AX205型,瑞士Mettler-Toledo公司;
超纯水仪:Integral 3型,法国Millipore公司;
数控超声波清洗器:KQ-700DV型,昆山市超声仪器有限公司;
无菌滤膜:0.2 μm;
移液器:100μL,1 mL;
卡托普利:批号100318-201105,含量99.5%,中国食品药品检定研究院;
卡托普利二硫化物:批号100319-201105,含量为100.0%,中国食品药品检定研究院;
卡托普利片:由A、B、C 3家生产企业提供,A企业有2个批次,规格均为25 mg;
乙腈:色谱纯,美国Fisher Scientific公司;
实验用水为自制超纯水(电阻率为18.25 MΩ·cm);
实验用其它试剂均为分析纯。
1.2 色谱条件
色谱柱:Water Acquity UPLC BEH C18色谱柱(2.1 mm×100 mm,1.7 μm);流动相:乙腈-水(体积比为25∶75,以磷酸调节pH为3.0),流速为0.2 mL/min;柱温:35℃;检测波长:215 nm;进样体积:5 μL。
1.3 溶液配制
1.3.1 对照品溶液
避光精密称取二硫化物12.51 mg,置于25 mL棕色量瓶中,加甲醇振摇溶解后定容,作为对照品储备液,于4℃冰箱避光保存。精密称取10.33 mg卡托普利样品于100 mL棕色量瓶中,精密加入3 mL二硫化物储备液,用流动相溶解定容,作为系统适用性试验溶液[16]。
1.3.2 供试品溶液
取样品10片称量,避光研细,精密称取细粉适量(相当于卡托普利25 mg),置于50 mL量瓶中,加入约25 mL流动相振摇10 min,用流动相定容后摇匀,备用。
2 结果与讨论
2.1 供试品溶液制备方法
优选了不同提取条件制备供试品溶液,采用不同提取方式(超声、振摇),不同振摇时间(5,10,15 min),最终确定振摇10 min,此条件下二硫化物基本提取完全,且振摇不会升温,比超声提取法更加温和,可避免不当前处理造成的正误差,方法简单,成本较低。
2.2 流动相
在流动相的筛选过程中,考察了甲醇-磷酸盐缓冲液、乙腈-磷酸盐缓冲液(1 g NaH2PO4溶解到1 L水中,磷酸调pH为3.0)、乙腈-水(以磷酸调节pH为3.0)等度洗脱。试验发现在相同配比、流速的条件下,甲醇洗脱体系的洗脱效率不如乙腈体系而压力却更高,采用水代替磷酸盐缓冲液色谱峰变化不大,故选择乙腈-水为流动相,利于保护色谱柱。流动相中有机相的比例变化对二硫化物的保留时间及其与主峰的分离度影响较大,乙腈含量为30%时,分离度低于4.0;降低乙腈含量至25%时,分离度达到6.0,满足药典要求;若再降低至20%,虽然分离度增至8.7,但出峰时间延长约1倍且理论塔板数降低约一半。故采用25%乙腈,出峰时间合适,分离度和理论板数较高,柱压较小。
2.3 流动相流速
在流动相为乙腈-水(体积比为25∶75)、柱温为35℃的条件下,比较了0.1,0.2,0.25,0.3 mL/min不同流速对色谱分离的影响。虽然二硫化物的保留时间随流速升高而缩短,但与主峰的分离度以及峰面积与理论板数都会降低,另外柱压较高,容易超限漏液,故最终选择流动相流速为0.2 mL/min。
2.4 色谱系统适用性
取对照品溶液、供试品溶液及稀释样品的流动相(作为空白样品溶液)各5 μL注入液相色谱仪,按外标法以峰面积计算二硫化物的含量,色谱图见图1。由图1可知,空白样品溶液色谱图中在被测组分峰位上没有吸收峰,故对测定无干扰。二硫化物保留时间约为4.2 min,约为主峰的2倍,与主峰的分离度大于6.0,单针运行5 min即可,理论踏板数按二硫化物计算不低于5 000,脱尾因子在0.9~1.0之间,峰形良好。
2.5 线性关系、检出限与定量限
移取二硫化物对照品储备液适量,加入流动相稀释配制成0.2,0.5,1,2,5,10,20 μg/mL的对照品标准溶液系列。分别取5 μL注入液相色谱仪,记录色谱图,以峰面积Y对各对照品浓度X(μg/mL)进行线性回归,得到的回归方程为Y=30 618.8X-1 230.2,相关系数r=0.999 95,表明二硫化物浓度在0.2~10 μg/mL的范围内与色谱峰面积呈良好的线性关系。
将0.2 μg/mL二硫化物对照品溶液用流动相逐级稀释后进样测定,得到二硫化物的检出限LOD(S/N=3)为0.1 μg/mL,定量限LQD(S/N=10)为0.2 μg/mL。

图1 空白样品(A)、混合对照品(B)、二硫化物对照品(C)和卡托普利片样品(D)的色谱图
2.6 精密度试验
取同一供试品溶液连续进样6次,峰面积测定结果见表1。由表1可知,二硫化物峰面积测定结果的相对标准偏差为0.65%,表明方法精密度良好。

表1 精密度测定结果
2.7 加标回收试验
取已知二硫化物含量的卡托普利片细粉6份(二硫化物含量为0.004 9 g/g),每份约80 mg(平均片重为83.4 mg),精密称定,置于50 mL容量瓶中。分别精密加入1 mL二硫化物储备液,加标量为0.5 mg,按1.2.2方法制备供试品溶液,依法测定并计算回收率,结果见表2。由表2可知,二硫化物平均回收率为98.9%(n=6),表明方法准确度良好。
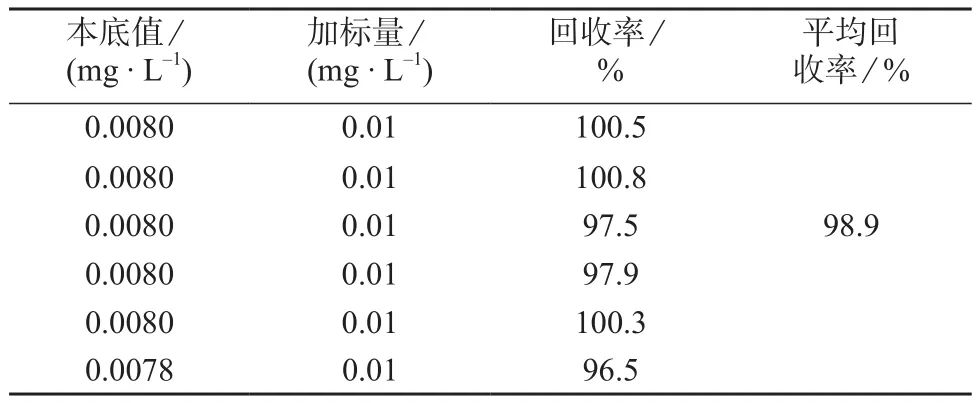
表2 二硫化物加标回收试验结果
2.8 样品测定
采用所建方法对来自3家企业的4批卡托普利片样品(A1,A2,B,C)进行测定,二硫化物质量浓度测定结果见表3。由表3可知,有一批检出二硫化物含量占标示量的3.8%,为不合格批次(药典规定二硫化物的限度为不得超过标示量的3.0%)。

表3 样品中二硫化物测定结果
3 结语
建立了超高效液相色谱法测定卡托普利二硫化物含量的方法,该法测定二硫化物保留时间稳定,测定结果准确、精密度高、回收率符合检测要求。与常规液相色谱法比较,超高效液相色谱的分析时间大幅缩短,分离效率高,重复性好,溶剂用量少。该法可作为实验室方法改进的参考依据,可用于卡托普利片的质量控制。
[1] 国家药典委员会.中国药典2010年版第一增补本[M].北京:中国医药科技出版社,2012:329.
[2] 蔡涛,任修文.对卡托普利片中卡托普利二硫化物检查项测定方法的改进[J].中国药事,2014,28(11): 1 252-1 256.
[3] 刘明理,马双成.药品快速检验技术研究及应用管理中需要加强的工作[J].中国药事,2012,26(8): 857-870.
[4] 石俊敏,管佳,张庆文,等.超高效液相色谱在药物分析中的应用[J].药物分析杂志,2008,28(9): 1 583-1 588.
[5] 李耿,孟繁蕴,杨洪军,等. UPLC法同时测定脑心通胶囊中5个成分的含量[J].药物分析杂志,2013,33(3): 414-418.
[6] 王丽娜,黄峻榕,张立,等.超高效液相色谱法测定婴幼儿配方奶粉中的叶黄素[J].色谱,2013,31(12): 1 228-1 231.
[7] 王瑞国,苏晓鸥,王培龙,等.超高效液相色谱法测定饲料中苯乙醇胺A[J].分析化学,2013,41(3): 389-393.
[8] 叶曦雯,彭燕,牛增元,等.超高效液相色谱-线性离子阱/静电场轨道阱高分辨质谱鉴别纺织品中禁用芳香胺及其异构体[J].色谱,2014,32(9): 1 005-1 012.
[9] 王点点,陈源,宋宁慧,等.超高效液相色谱法测定稻田土壤及水中的噻虫啉与氟虫双酰胺残留[J].分析测试学报,2013,32(1): 138-142.
[10] 孟鹏,郑宝东.超高效液相色谱法快速并同时检测金柑中柠檬苦素和诺米林[J].中国食品学报,2013,13(2): 177-181.
[11] 彭涛,王超,吕怡兵,等.超高效液相色谱法快速检测地表水中丁基黄原酸[J].中国环境监测,2013,29(2): 65-68.
[12] 王星,池玉梅,康安.超高效液相色谱-串联质谱法定性和高效液相色谱法定量分析天南星中氨基酸成分[J].色谱,2014,32(12): 1 326-1 332.
[13] 龙泽荣,徐微微,鹿毅,等.分子印迹固相萃取-超高效液相色谱法测定河水中的碱性橙Ⅱ[J].理化检验:化学分册,2014,50(8): 937-943.
[14] 李慧勇,谭建华,郭长虹,等.化妆品中黄芩苷与白鲜碱的超高效液相色谱检测及质谱确证[J].分析测试学报,2014,33(12): 1 399-1 403.
[15] 崔思娇,罗洁,马天成,等.酸枣仁的超高效液相色谱指纹图谱[J].中国药学杂志,2013,48(7): 509-511.
[16] 国家药典委员会.中国药典2010年版第二部[M].北京:中国医药科技出版社,2010: 130.
我国已制定492项食品安全国家标准
根据国务院食品安全委员会办公室等18个部门联合下发的通知,2015年全国食品安全宣传周活动已正式拉开帷幕。在以“尚德守法,全面提升食品安全法治化水平”为主题的全国食品安全宣传周主场活动上,记者获悉,我国迄今已制定公布492项食品安全国家标准,基本覆盖保障食品安全的主要指标万余项。
作为本次宣传周主场活动的重头戏,在当天的分论坛——“聚焦新修《食品安全法》”上,食品药品监管总局相关负责人指出,作为此次修法重要的增加条款之一,网络食品交易的相关规定填补了对食品生产、流通、餐饮服务和食用农产品销售等各个环节存在的网络交易监管制度空白,将保证消费者在互联网食品交易中所产生的交易纠纷,能够迅速得以解决。
1 强化食品安全统一治理
全国人大高票通过新修订的食品安全法,进一步明确了预防为主、风险管理、全程控制、社会共治的原则,着力构建最严格的覆盖全过程的监管制度。
新法从104条增加到154条,新增50条,对原有70%的条文进行了实质性修改;法律文本从1.5万字增加到3万字;法律责任从15条增加到28条。这部被称为“史上最严”的食品安全法,标志着我国食品安全工作在法治化轨道上又迈出了重大步伐。
2 体现刑事责任追究优先
公安部治安管理局打假处处长许成磊也认为,新法充分体现了“最严厉的处罚”。而其中最突出的表现,就是对一些严重危害人体健康的食品安全违法行为体现了“刑事责任追究优先”。许成磊说,新食品安全法突出“刑事责任优先”,比如第123条规定经营病死、死因不明的禽、畜、兽、水产动物肉类及其制品的,原则上即构成刑法第143条规定的生产销售不符合安全标准的食品罪,要予以刑事追究。
新法还首次规定对危害较大、尚未构成犯罪的7种违法情形由公安机关给予行政拘留处罚。许成磊说,此次新修订食品安全法规定了7种危害相对较大的食品安全违法情形在达不到刑事追究的情况下,情节严重的,在给予吊销许可证的同时,可由公安机关对直接负责的主管人员和其他直接责任人员处5日以上15日以下拘留。
3 新增互联网食品交易监管
随着我国电子商务的迅猛发展,网购已经成为居民日常消费的主要方式之一。在此背景下,新食品安全法对互联网食品交易的规定,也是此次修法的一大关注点,是重要的增加条款之一。
食品药品监管总局食品监管二司司长张靖说,网络食品交易存在一定风险,当前互联网食品经营监管面临一些难以解决的问题,既有产业政策跟不上的问题,也有监管能力跟不上的问题。因此,将于2015年10月1日实施的新版食品安全法,对网络食品经营新增了制度性规定。
张靖认为,新法通过规定互联网食品交易第三方平台三类义务,使互联网食品经营中食品安全、消费者权益保护等问题能够落实责任承担者,确保互联网食品交易的安全,同时也保证消费者在互联网食品交易中所产生的交易纠纷,并能够迅速得以解决。
(中国分析计量网)
DNA分子检测方法成为国标
国家标准化管理委员会批准国家标准《农作物种子检验规程真实性和品种纯度鉴定》(GB/T3543.5-1995)第1号修改单,其中规定品种真实性或身份鉴定允许采用DNA分子检测方法,这为快速准确打击品种假冒侵权违法行为提供了有力依据,该修改自2015年7月1日起实施。
修改单中增加6.2.4 DNA分子检测方法,具体内容:品种真实性验证或身份鉴定,允许采用简单重复序列(简称SSR)和单核苷酸多态性(简称SNP)分子标记方法。检测采用抽取有代表性的检测样品与标准样品、DNA指纹数据库比较的方式。同时,修改单还将6.4条中“田间小区种植是鉴定品种真实性和测定品种纯度的最为可靠、准确的方法”修改为“田间小区种植是鉴定品种真实性和测定品种纯度的可靠方法之一。”
(仪器信息网)
Determination of Captopril-Disulphide in Captopril Tablets by Ultra-Performance Liquid Chromatography
Tan Jinfeng1, Ma Yunyun2
(1. Weifang Environmental Monitoring Center, Weifang 261041, China; 2. Weifang Institute for Food and Drug Control, Weifang 261041, China)
Ultra-performance liquid chromatographic method for the determination of captopril-disulphide in captopril tablets was established. The method was performed on a Waters Acquity UPLC BEH C18analytical column and the mobil phase consisted of acetonitrile-H2O (pH 3.0) with flow rate of 0.2 mL/min. The injection volume was 5 μL and the content was quantited by the area of the disulphide peak with the standard curve. The concentration of captopril-disulphide was linear with the peak area in the range of 0.2-10 μg/mL,r=0.999 95. The relative standard deviation of area detection results was 0.65% (n=6). The average recoveries was 98.9%, the peaks of captopril and disulphide were well isolated and the chromatographic separation degree was more than 6.0. The method was used for detecting four batches of samples, and the results were in the range of 0.3-3.8%. Compared with routine liquid chromatography,this method has high separation and good repeatability,which can be used for the quality control of captopril.
UPLC; captopril-disulphide; content determination; quality control; rapid test
O657.7
:A
:1008-6145(2015)04-0036-04
10.3969/j.issn.1008-6145.2015.04.011
联系人:谭金峰;E-mail: tanjinfeng@163.com
2015-04-14
