具有特殊结构和电子性质的PdAu/Al2O3催化剂上蒽醌加氢反应性能
2015-01-04何志远管永川张金利天津大学化工学院天津300072
韩 优 何志远 管永川 李 韡 张金利(天津大学化工学院,天津300072)
具有特殊结构和电子性质的PdAu/Al2O3催化剂上蒽醌加氢反应性能
韩 优 何志远 管永川 李 韡 张金利*
(天津大学化工学院,天津300072)
通过改变Pd和Au的负载顺序合成了一系列具有不同结构和电子性质的PdAu双金属催化剂,并用于蒽醌加氢反应.其中通过先负载Au后负载Pd的顺序制得的Pd/Au/Al2O3催化剂,其加氢效率可高达14.27 g·L-1. X射线衍射、透射电子显微镜、H2程序升温还原和X射线光电子能谱等分析表征结果显示,Pd/Au/Al2O3催化剂中分散在Au颗粒表面的Pd纳米颗粒具有独特的爆米花结构,其表面零价态的单质Pd含量最多,而这种表面零价态的单质Pd是蒽醌加氢反应中的关键活性组分.此外,Au的加入可有效抑制副反应的发生,减少降解产物的生成,从而大大提高了催化选择性.
负载顺序;PdAu双金属;蒽醌加氢;降解产物
1 Introduction
Catalytic hydrogenation of 2-ethylanthraquinone(EAQ)into 2-ethylanthrahydroquinone(EAQH2)is the key reaction in the industrial synthesis of H2O2.1,2In this method,EAQ is hydrogenated to EAQH2in the presence of metal catalyst,and then EAQH2is oxidized by O2to yield H2O2with regeneration of the starting EAQ.The target product EAQH2formed in the quinonehydroquinone stage undergoes further hydrogenation to givevarious products through two reaction pathways.The first reaction route consists of the successive saturation of aromatic ring to generate 2-ethyl-5,6,7,8-tetrahydro-9,10-anthrahydroquinone (H4EAQH2)and 2-ethyl-1,2,3,4,5,6,7,8-octahydro-9,10-anthrahydroquinone(H8EAQH2).The second route is the hydrogenolytic cleavage of C―O bonds to give 2-ethylanthrone(EAN) as the main product.Among them only H4EAQH2can be oxidized by O2to generate H2O2and its corresponding 2-ethyl-5,6,7,8-tetrahydro-9,10-anthraquinone(H4EAQ)can also be used in the production cycle of H2O2.Thus EAQ and H4EAQ are called as“active quinones”and other hydrogenation products are considered as“degradation products”.3-6Limiting degradation products formed in the anthraquinone process is one of the priority issues of process improvement,since the formation of degradation products substantially reduces the amount of active quinones.7
©Editorial office ofActa Physico-Chimica Sinica
Drelinkiewicz et al.5,8,9carried out pioneering work to investigate feasible method to limit the formation of degradation products on Pd catalysts.They reported that alkali modifiers(Li,Na, K,Cs)could accelerate the rate of the first quinone-hydroquinone stage whereas reduce the rate of reactions in“deep hydrogenation”stage.The suppression of“deep hydrogenation”stage was ascribed to reduced acidity of catalysts by alkaline promoters.5They also coated SiO2with polyaniline(PANI).The prepared Pd/ PANI(SiO2)catalyst exhibited high reactivity to phenyl ring saturation and strong inhibition of degradation reactions compared with conventional Pd/SiO2catalyst.Such advantageous catalytic properties may be related with the weakening of the strength of hydrogen bonding and hydrophobic character of polymer.8Modification of the Al2O3support with Na2SiO3solution was another effective way to limit hydrogenolytic reactions producing degradation products and enhance the maintenance of catalysts activity during the hydrogenation experiment.The observed phenomenon showing a profitable role of silica species may be interpreted by modification of adsorption properties of the surface towards anthraquinone molecules.9Li et al.3,10improved preparation methods ofAl2O3support used in EAQ hydrogenation.They prepared spherical Al2O3,SiO2-Al2O3using conventional oil-drop (OD)method,and Al2O3using the separate nucleation and aging steps(SNAS)method.The order of average hydrogenation efficiency and selectivity to active quinones was Pd/Al2O3(SNAS) (10.9 g·L-1,97%)>Pd/SiO2-Al2O3(OD)(10.3 g·L-1)>Pd/Al2O3(OD)(8.6 g·L-1,93%).The improved performance can be ascribed to the modified pore structure of support,which can increase the dispersion of Pd and decrease the diffusion resistance.
These researches mainly focused on the modification of supports.Few attempts have been made to limit the formation of degradation products through controlling the catalytic chemistry of Pd based on catalyst design principles.A potential method to improve catalytic performance is incorporation of a second metal into the Pd catalyst.11-13Au has been found to enhance the activity of Pd significantly by the pioneer work of Hutchings et al.14-16Bimetallic PdAu catalysts have been reported to be active for a variety of selective hydrogenation reactions.Yang et al.17reported that bimetallic PdAu catalyst showed a higher activity and selectivity than Pd catalyst in hydrogenation of phenol to cyclohexanone,conversion increased from 15%to 97.5%and selectivity increased from 94.6%to 96.6%.Panpranot et al.18found that Au in bimetallic PdAu catalyst for 1-heptyne hydrogenation could promote electrons for Pd,which greatly promoted the second-step hydrogenation of 1-heptene to heptane.Yang et al.19also found the addition of Au to Pd catalyst favorably promoted the hydrogenation of cinnamaldehyde with conversion rate improved for three times.The selectivity to hydrocinnamal dehyde was also increased from 85%to 90%.Ouyang et al.20revealed that the role of Au in bimetallic PdAu catalyst for the direct synthesis of H2O2from H2and O2was to isolate single Pd sites to facilitate the dissociation of H2but unfavorable for the dissociation of O2.This promotion effect dramatically enhanced the selectivity to H2O2and reduced the decomposition of H2O2.Based on these encouraging findings, incorporation of Au into Pd/Al2O3catalyst becomes a meaningful and feasible method to improve its catalytic activity and selectivity for the hydrogenation of EAQ.
In our present work,a combination of impregnation(IM)and deposition-precipitation(DP)methods was used to prepare the bimetallic PdAu/Al2O3catalysts with high performance for the EAQ hydrogenation reaction.In an attempt to establish a relationship between the catalytic performance and their structural and electronic properties,a systematic investigation of the structural and electronic properties of PdAu/Al2O3catalysts with different preparation methods was carried out via various analytic techniques,such as N2adsorption-desorption,X-ray diffraction (XRD),transmission electron microscopy(TEM),CO chemisorption,hydrogen temperature program reduction(H2-TPR),and X-ray photoelectron spectroscopy(XPS).
2 Experimental
2.1 Catalyst preparation
In this work,HAuCl4·3H2O(99.99%Sigma-Alddrich)was used as gold precursor and PdCl2(99.99%Sigma-Alddrich)as palladium precursor.Pseudoboehmite(99%CNOOC Tianjin Chemical Research Institute)was calcined in air at 550°C for 4 h to obtain γ-Al2O3.The particle size was remained between 0.02 and 0.10 mm.γ-Al2O3was used as a support for preparing three bimetallic PdAu samples with the same Pd(0.3%,w,mass fraction,the same below)andAu(0.28%)loadings at a mole ratio of 2:1 but prepared with three different procedures as follows.
(i)The catalyst(Pd/Au/Al2O3)was prepared by depositing the two metals separately using different techniques(i.e.,Pd by IM and Au by DP).Au was deposited using DP by suspending the support in a HAuCl4aqueous solution.The pH was adjusted to 7 by dropwise addition of a 1 mol·L-1NaOH solution(96%,Tianjin Guangfu Fine Chemical Research Institute).Then,the material was vigorously mixed for 2 h and maintained standing at 45°C for 12 h.The obtained sample was dried at 120°C overnight and calcined in static air at 550°C for 4 h.In the second step,theobtained Au/Al2O3was impregnated with a PdCl2aqueous solution.Then,the obtained sample was subjected to the same treatment conditions described above.
(ii)Another catalyst sample(Au/Pd/Al2O3)was prepared by inverting the deposition order where the Pd was deposited by IM in the first step followed byAu loading using a DP method in the second step.
(iii)The third catalyst(Pd-Au/Al2O3)was prepared by co-DP. Astirred slurry of γ-Al2O3in a PdCl2and HAuCl4aqueous solution was adjusted to pH 7 by dropwise addition of a 1 mol·L-1NaOH sodium hydroxide solution.Then the obtained sample was subjected to the same treatment conditions described above.
For comparison,a monometallic Pd catalyst(Pd/Al2O3)was prepared by IM,and a monometallic Au catalyst(Au/Al2O3)was synthesized by DP.The detailed procedures were the same as those used for the bimetallic catalysts.In all of these catalysts,we kept the method of Pd(or Au)addition to the catalyst consistent, i.e.,Au by DP and Pd by IM(except the Pd-Au/Al2O3because of the procedure limitation).All of the catalysts were prepared by reducing the sample for 2 h under a H2flow at 350°C with a rate of 5°C·min-1.
2.2 Catalyst characterization
N2adsorption-desorption isotherms were determined at-196°C using a ASAP 2000 analyzer(Micromeritics,America).Inductively coupled plasma-atomic emission spectrometry(ICP-AES) was carried out using an Iris advantage device(Thermo Jarrel Ash,America).XRD patterns were measured from 10°to 90°(2θ) using a Bruker D8Advance diffractometer equipped with a Si(Li) solid-state detector(SOL-X)and a sealed tube providing Cu Kαradiation(Bruker,Germany).TEM and scanning transmission electron microscopy(STEM)analysis were carried out using a JEOL JEM2010 microscope under an accelerating voltage of 200 kV(JEOL,Japan).This instrument includes an EDAX X-ray energy dispersive spectrometer(EDS)and a JEOL high-angle annular dark-field(HAADF)detector.The XPS spectra were recorded using an Axis Ultra DLD spectrometer employing a monochromated Al-KαX-ray source(hν=1486.6 eV),hybrid (magnetic/electrostatic)optics,a multi-channel plate,and delayline detector(DLD)(Kratos,Britain).All the binding energies were referenced to the C 1s peak at 284.6 eV.H2-TPR was performed with a TPDRO1100 instrument(Thermo Jarrel Ash, America):a sample of 50 mg was heated from room temperature to 900°C at a heating rate of 10°C·min-1in a 10.0%H2-Ar mixture flowing at a rate of 20 mL·min-1.A TPDRO1100 instrument was used to perform pulse chemisorption to determine the CO uptake(Thermo Jarrel Ash,America).The catalysts were reduced at 550°C respectively for 1 h and then cooled to room temperature in He.Pulse CO chemisorption was performed using 500 μL pulse of 10%CO/He in a He carrier gas.A 1:1 CO/Pd molar ratio was assumed to determine the Pd surface content of the catalysts.
The N2adsorption-desorption,ICP,H2-TPR,and CO chemisorption investigations were performed with fresh catalysts,and the XRD,TEM,and XPS investigations were performed using the reduced catalysts.
2.3 Catalytic performance test
The hydrogenation experiments were carried out in an autoclave at 0.3 MPa and 60°C.The working solution was prepared by dissolving 120 g of solid EAQ(98%,TCI)in 1 L of a mixed solvent of trioctyl phosphate(98%,TCI)and trimethylbenzene (98%,TCI)with the volume ratio of 0.33.60 mL of the working solution was mixed with 1.20 g of catalyst,and then,the mixture was hydrogenated in a H2atmosphere for 30 min.After the hydrogenation reaction,the solution was instantaneously centrifugally separated with a rotating speed of 3000 r·min-1for 15 min to remove the solid catalyst.Then,2 mL of the catalyst-free solution was placed into 20 mL of deionized water,and the mixture was oxidized by oxygen at room temperature for 30 min in a separating funnel.2 mL of dilute phosphorous acid was added to the deionized water to prevent H2O2decomposition during the oxidation reaction.After the oxidation reaction,a H2O2aqueous solution was obtained from the sublayer solution of the separating funnel.The H2O2content was analyzed by titration with a KMnO4solution.Prior to the titration,5 mL of a 20%(w)sulfuric acid solution was added to the H2O2solution.The catalytic activity is expressed by the following simplified equation:3where B is the hydrogenation efficiency(g·L-1),which is defined as the mass of H2O2formed per volume working solution,C is the KMnO4solution concentration(mol·L-1),V0is the KMnO4solution volume(mL),and V is the H2O2solution volume(mL).

A high performance liquid chromatograph(HPLC)equipped with a C18 separation column and UV detector was used to analyze the concentrations of EAQ and H4EAQ,which were denoted as ct(EAQ)and ct(H4EAQ),respectively,at t=30 min in the reoxidized working solution.The mobile phase was a mixture of methanol and water with a volume ratio of 80:20.The wavelength of the ultraviolet radiation was 245 nm.The sum of the EAQ and H4EAQ concentrations in the solution was smaller than the initial concentration of EAQ(i.e.,c0(EAQ)).This difference was assumed as cumulative of the degradation products(cD)given by a mass balance:4,6

3 Results and discussion
3.1 Structural properties
3.1.1 N2adsorption-desorption
The chemical compositions and textural properties of the calcined catalysts are summarized in Table 1.The N2adsorptiondesorption isotherm for the γ-Al2O3(Fig.S1,Supporting Information)showed that it was a type IV isotherm with hysteresis loop of mesoporous materials.The bare γ-Al2O3exhibited mesoporous structure with SBET=190 m2·g-1,Vp=0.56 cm3·g-1,Dp=11.7 nm.When the support was loaded with metal,its surface area and total pore volume(for example,Pd/Au/Al2O3SBET=174 m2·g-1,Vp=0.54 cm3·g-1)descended.This was caused by some Pd or Au complexes partially blocking the micropores during the calcination process.
The Pd andAu amounts of the catalysts were analyzed by ICP. As listed in Table 1,the Pd andAu amounts found in the catalysts were slightly lower than the theoretical value(i.e.,0.30%for Pd and 0.28%for Au),but their mole ratio in the bimetallic catalysts equalled to the nominal value of 2:1.

Table 1 Chemical composition and textural properties of the calcined catalysts and bare support
3.1.2 XRD
The metal crystallites of the catalysts were characterized using XRD.As shown in Fig.1,peaks located at 2θ=38.1°,44.3°,and 77.6°were indexed to the(111),(200),and(311)planes of cubic Au metal.19The XRD characteristic peaks corresponding to Pd,i.e., (111)at 40.1°and(200)at 46.6°,were not detected in any of the catalysts,which might be due to the overlapping with the broad feature from theAl2O3support,(222)at 39.5°and(400)at 45.9°.21Table 2 presents the Au particle size data calculated using the Scherrer equation.The fairly large particle size(16.3-21.1 nm) was due to the calcination at a high temperature(550°C)in air.22Because the average pore diameter of the bare support was 11.7 nm,these particles could not enter into the Al2O3pores and were located on the external surface.23

Fig.1 XRD patterns for theAl2O3,Au/Al2O3,Pd/Al2O3,Pd/Au/ Al2O3,Au/Pd/Al2O3,and Pd-Au/Al2O3after reduction

Table 2 Specifications of several catalysts prepared using different methods
3.1.3 TEM
In order to gain a better understanding of the microstructure of the PdAu particles,the samples were observed using STEMHAADF and TEM.TheAu particles with an average size of 13.9 nm were clearly visible in the TEM images ofAu/Al2O3in Fig.S2 (Supporting Information).TEM results for Pd/Al2O3indicated that Pd was better dispersed on theAl2O3support with an average size of 6.2 nm.For Pd/Au/Al2O3,as shown in Fig.2(a),PdAu particles dispersed homogeneously on support,with an average particle size of 15.2 nm.The HRTEM image(Fig.2(b))showed that these Pd nanoclusters with popcorn structure were dispersed on the Al2O3supported Au particles.The STEM-HAADF and linescanning EDS results shown in Fig.2(c)and 2(d)further confirmed the observation from the HRTEM.The Pd particles were 4-5 nm and theAu particle was~15 nm.
For the Au/Pd/Al2O3,particles with an average size of 14.9 nm were observed(Fig.3(a)).In a random selection of these bimetallic particles(Fig.3(b)),three Pd particles were surrounded with Au. The STEM-HAADF and line-scanning EDS(Figs.3(c)and 3(d)) indicated that the partially coverage of Pd surface by Au.24In our present study,Au particles loaded in the second step could melt during the calcination process.25TheseAu species incorporated Pd particles that were near from each other into larger aggregates.
A totally different morphology was observed for the Pd-Au/ Al2O3catalyst,which exhibited a vast quantity of larger bimetallic particles with an average size of 25.8 nm(Fig.4(a)).The HRTEM image(Fig.4(b))showed that largeAu particle was covered by Pd. The STEM-HAADF and line-scanning EDS shown in Fig.4c and 4d further confirmed theAucore-Pdshellstructure was formed in Pd-Au/Al2O3catalyst.The core-shell structure of supported bimetallic PdAu particles with relative large size(>30 nm)prepared by the same co-IM method has also been reported by other research groups.14,25-28On the premise of this structure,the Au particle size (14.9 nm)based on XRD results only reflected the size of Au core,which was in accordance with size observed by line-scanning EDS(15-17 nm).The Pd particle size(25.6 nm)based on CO chemisorption results(in Table 2)reflected the size of these Aucore-Pdshellparticles,which was in accordance with size observed by TEM(25.8 nm).
3.1.4 CO chemisorption
CO chemisorption was performed to determine the Pd dispersion and particle size.26The corresponding results are shown in Table 2.The dispersion of Pd increased in the following order: Pd/Au/Al2O3>Pd/Al2O3>Au/Pd/Al2O3>Pd-Au/Al2O3.The calculated size of Pd nanoparticle in Pd/Au/Al2O3catalyst was in agreement with that observed by line-scanning EDS results.The popcornstructure of Pd nanoparticles formed on the supportedAu particles improved the dispersion of Pd.For Au/Pd/Al2O3,the lower dispersion compared with Pd/Al2O3was due to the coverage by Au. And the lowest dispersion of Pd-Au/Al2O3was due to the formation of large particles.

Fig.2 (a)TEM image,(b)HRTEM image,and(c)STEM-HAADF image of Pd/Au/Al2O3catalyst; (d)line-scanning EDS of the metal particle indicated with a line in STEM-HAADF image

Fig.3 (a)TEM image,(b)HRTEM image,and(c)STEM-HAADF image ofAu/Pd/Al2O3catalyst; (d)line-scanning EDS of the metal particle indicated with a line in STEM-HAADF image

Fig.4 (a)TEM image,(b)HRTEM image,and(c)STEM-HAADF image of Pd-Au/Al2O3catalyst, (d)line-scanning EDS of the metal particle indicated with a line in STEM-HAADF image
3.2 Electronic properties
3.2.1 H2-TPR
To further understand the interaction between Pd and Au,the reducibility of the unreduced samples was investigated by H2-TPR,and the results are shown in Fig.5.The Au/Al2O3catalyst exhibited a broadened H2consumption peak in the temperature range of 170-210°C,which may be due to the reduction ofAu3+.19For the Pd/Al2O3catalyst,peaks observed at 130,150,and 375, 460°C were due to the reduction of surface PdO,Pd2O and subsurface PdO,Pd2O,respectively.29For the bimetallic PdAu catalysts,all of the reduction peaks for the Pd species shifted to lower temperatures,and the extent of the shift for Pd/Au/Al2O3and Au/Pd/Al2O3was larger than that for Pd-Au/Al2O3.In addition,the first reduction peak that appeared at 100°C for Pd/Au/Al2O3was much sharper than that of Au/Pd/Al2O3,which further demonstrated that the Pd in Pd/Au/Al2O3possessed a relatively uniform particle size.It should be noted that the dissociative H2adsorption capacity of the metal catalyst is related to the electronic structure of the metal.19The results of H2-TPR suggested that the addition ofAu have modified the electronic properties of Pd.

Fig.5 H2-TPR curves forAl2O3,Au/Al2O3,Pd/Al2O3, Pd/Au/Al2O3,Au/Pd/Al2O3,Pd-Au/Al2O3
3.2.2 XPS
To obtain additional insight into the electronic relationship between the Pd and Au species in the bimetallic PdAu catalysts, the chemical states of Pd andAu were investigated by XPS.Prior to the measurements,all of the catalysts were reduced in H2and maintained in a vacuum desiccator.The test samples were prepared in a glovebox in order to avoid the contact with air.The Pd 3d andAu 4f spectra of the catalysts are shown in Fig.6.Ashift of ca 0.1-0.8 eV was observed toward high binding energy compared to the monometallic Pd for the three bimetallic PdAu catalysts,which was due to the charge transfer betweenAu and Pd.19The presence of Pd2+in all of these catalysts resulted from the incomplete reduction of some surface oxide layer of PdO.18

Fig.6 XPS spectra of Pd 3d(a)andAu 4f(b)in the different reduced catalysts
The fraction of Pd species on the surface was roughly represented by the peak-fitting method.The XPS-derived atomic ratios are listed in Table 3.It is important to note that the addition ofAu elevated the ratio of Pd0in these bimetallic PdAu samples compared to that in the pure Pd/Al2O3catalyst,which was consistent with the results of other researchers.30,31Further analysis indicated that the percentage of Pd0on the surface increased in the following order:Pd/Au/Al2O3>Au/Pd/Al2O3>Pd/Al2O3>Pd-Au/Al2O3, and the percentage of Au0on the surface decreased in the same order.These results implied that the addition of Au benefited the reduction of Pd2+to metallic Pd or the protection of Pd0from being oxidized to Pd2+.In addition,the Au0species can act as an electronic promoter for Pd.

Table 3 Surface atomic ratios(%)of the catalysts after reduction based on XPS
3.3 Mechanism of loading sequence effect on the structural and electronic properties of bimetallic catalysts
Based on the characterization results,the schematic diagrams at the top-left corner of Figs.2(b),3(b),and 4(b)showed the structures of the reduced bimetallic PdAu/Al2O3catalysts prepared via different loading sequences.For Pd/Au/Al2O3,Pd was subsequently loaded on the Au particles to form a popcorn structure with a uniform particle size distribution and an improved Pd species dispersion.In addition,the subsequently loaded Pd nanoclusters on the Au particles might inhibit the aggregation of the Au particles during the second calcination step due to its higher melting point.25Therefore,more Pd atoms were exposed on the surface of the catalyst.For Au/Pd/Al2O3,Pd was partially covered by Au,thus it contained a lower content of surface Pd. The Pd-Au/Al2O3sample exhibited larger bimetallic particles with Aucore-Pdshellstructure.Due to the low dispersion,much fewer surface Pd atoms were exposed.
The different size distributions and morphologies of the supported PdAu catalysts led to different interactions betweenAu and Pd.The results of a theoretical study32indicated that the Au adatom was basically neutral on the Al2O3support,while the Pd adatom could provide a significant amount of charge transfer to the Al2O3surface,which could explain the easy oxidation of Pd. When Au is deposited first followed by Pd loading to form the unique popcorn structure in the Pd/Au/Al2O3catalyst,a high content of Pd was formed on theAu surface rather than theAl2O3surface,which prevented the electron transfer from Pd to theAl2O3support and maintained more Pd in the metallic state.In addition, the popcorn structure of the PdAu nanoparticles in Pd/Au/Al2O3, which were uniform and small in size,increased the contact area between Au and Pd,which led to a shortcut for electron transfer fromAu to Pd and reduction of Pd2+to metallic Pd.Therefore,the content of metallic surface Pd in Pd/Au/Al2O3was the highest among the three bimetallic PdAu catalysts prepared with different loading sequences.
3.4 Catalytic performance
The catalytic performance of the bimetallic PdAu/Al2O3catalysts is shown in Table 4.A negligible yield(1.11 g·L-1)wasobtained for Au/Al2O3under the given reaction condition,implying that Au was inactive for the EAQ hydrogenation reaction. OnceAu was added to the Pd/Al2O3,the Pd/Au/Al2O3(14.27 g·L-1) and Au/Pd/Al2O3(12.79 g·L-1)catalysts exhibited higher hydrogenation efficiency than Pd/Al2O3(12.04 g·L-1),especially for the Pd/Au/Al2O3catalyst.The hydrogenation efficiency increased in the following order:Pd/Au/Al2O3>Au/Pd/Al2O3>Pd/Al2O3>Pd-Au/Al2O3.This order was consistent with the surface Pd0content in these catalysts,which indicated that surface Pd0was a key active component for the EAQ hydrogenation reaction.The hydrogenation efficiency was 14.27 g·L-1for the Pd/Au/Al2O3catalyst.The hydrogenation efficiencies of different Pd/Al2O3catalysts measured by Li et al.3,10,33,34in a fixed bed reactor were 8.0,8.5,10.2,and 10.9 g·L-1.The catalytic performances of industrial catalysts were also evaluated under the same condition as our prepared catalysts and the corresponding data were also listed in Table 4.As determined by ICP analysis,the Pd amounts found in the industrial catalyst-1 and industrial catalyst-2 were 0.6%, 1.8%,respectively.Compared with these two industrial catalysts, the Pd/Au/Al2O3was highly active but with a much lower Pd loading.The hydrogenation efficiency based on the mass of Pd was calculated and increased in the following order Pd/Au/Al2O3(4757 g·L-1·g-1)>Pd/Al2O3(4013 g·L-1·g-1)>industrial catalyst-1 (1933 g·L-1·g-1)>industrial catalyst-2(712 g·L-1·g-1).
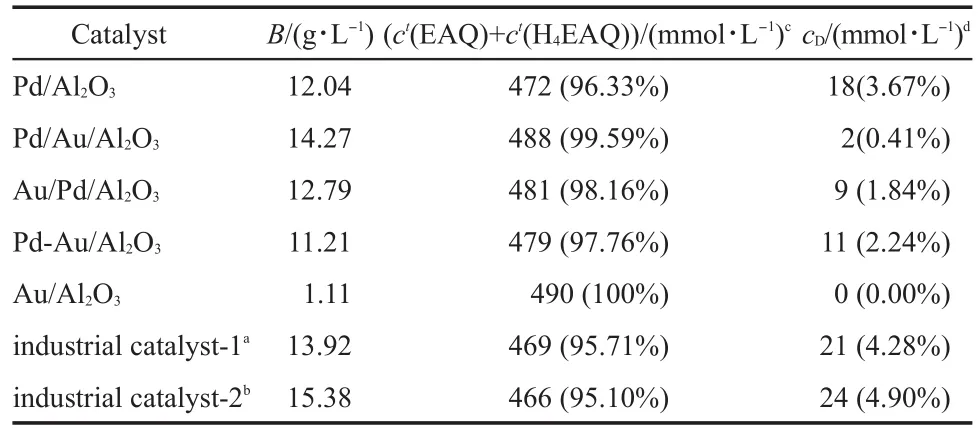
Table 4 Catalytic activities for EAQ hydrogenation with different catalysts and the composition of the re-oxidized working solution
In the anthraquinone process,only EAQ and H4EAQ are called“active quinones”because they can be used in the H2O2production cycle.Other hydrogenation products are considered as“degradation products”.10Because the degradation products that formed in the hydrogenation/oxidation process cycle will cause a loss of active quinones,the amount of active quinones is an important parameter in H2O2production using the anthraquinone method.The amount of active quinines(ct(EAQ)+ct(H4EAQ))and degradation products(cD)in the re-oxidized working solution are also listed in Table 4.It clearly shows that the amount of degradation products decreases via adding Au into the Pd/Al2O3catalyst,demonstrating that the addition of Au can effectively suppress the side reactions.The Pd/Au/Al2O3catalyst generated the fewest degradation products among all the catalysts in the first hydrogenation/oxidation process cycle.Its selectivity to active quinones was also much higher than the industrial catalysts.Table 5 presents the catalytic performance results for the optimal Pd/Au/ Al2O3catalysts at various mole ratios of Pd and Au.We kept the Pd loading constant(0.3%)and changed Pd/Au mole ratio by altering theAu loading.The catalytic performance results in Table 5 further demonstrated that the addition of Au could keep the catalytic selectivity at a high level(>99%)via suppressing the side reactions.Futhermore,the loading amount of Au could affect the catalytic activity to some extent,and the best mole ratio for Pd/Au/ Al2O3catalyst was 2:1.
Two types of compounds have been suggested for the composition of degradation products.One consists of the successive saturation of phenyl ring,such as H8EAQH2.The second route is the hydrogenolytic cleavage of C―O bonds,such as EAN.The hydrogenation of the second aromatic ring with formation of H8EAQH2was observed only after complete transformation of quinone system in EAQ to the EAQH2.35So the degradation products listed in Tables 4 and 5 mainly consist of hydrogenolysisderiving products.As a matter of fact,the side reactions comprising the hydrogenolytic cleavage of C―O bonds were strongly limited by addition ofAu.

Table 5 Catalytic activities for the optimal Pd/Au/Al2O3catalysts at various mole ratios of Pd andAu and the composition of the re-oxidized working solution
4 Conclusions
Pd/Au/γ-Al2O3catalyst prepared by loading Pd on Au particles deposited on γ-Al2O3support showed superior catalytic performance(i.e.,B=14.27 g·L-1,selectivity>99%)in the EAQ hydrogenation reaction.The morphology of the sample could be depicted as small,monodispersed Pd nanoparticles distributed on supported Au particles.Moreover,the electronic promotion effect of Au helped to keep more metallic Pd on the catalyst surface, which was the key active component for the reaction of EAQ hydrogenation.The addition of Au was found to improve the selectivity to active quinones.These conclusions not only provide the basis for the design of novel catalysts used in the industrial anthraquinone process,but also are helpful in the development of supported bimetallic PdAu catalysts for related reactions.
Supporting Information:The N2adsorption-desorption isotherms of γ-Al2O3and the prepared catalysts,and the TEM images for Pd/Al2O3and Au/Al2O3have been included.This information is available free of charge via the internet at http://www. whxb.pku.edu.cn.
(1) Campos-Martin,J.M.;Blanco-Brieva,G.;Fierro,J.L.G. Angew.Chem.Int.Edit.2006,45,6962.
(2) Samanta,C.Appl.Catal.A 2008,350,133.doi:10.1016/j. apcata.2008.07.043
(3) Feng,J.T.;Wang,H.Y.;Evans,D.G.;Duan,X.;Li,D.Q.Appl. Catal.A 2010,382,240.doi:10.1016/j.apcata.2010.04.052
(4) Drelinkiewicz,A.;Waksmundzka-Gora,A.J.Mol.Catal.A: Chem.2006,246,167.doi:10.1016/j.molcata.2005.10.026
(5) Kosydar,R.;Drelinkiewicz,A.;Lalik,E.;Gurgul,J.Appl. Catal.,A 2011,402,121.doi:10.1016/j.apcata.2011.05.036
(6) Kosydar,R.;Drelinkiewicz,A.;Ganhy,J.P.Catal.Lett.2010, 139,105.doi:10.1007/s10562-010-0413-1
(7) Chen,Q.L.Chem.Eng.Process 2008,47,787.doi:10.1016/j. cep.2006.12.012
(8) Drelinkiewicz,A.;Waksmundzka-Góra,A.;Makowski,W.; Stejskal,J.Catal.Commun.2005,6,347.doi:10.1016/j. catcom.2005.02.009
(9) Drelinkiewicz,A.;Kangas,R.;Laitinen,R.;Pukkinen,A.; Pursiainen,J.Appl.Catal.A 2004,263,71.doi:10.1016/j. apcata.2003.12.010
(10) Tang,P.G.;Chai,Y.Y.;Feng,J.T.;Li,Y.;Li,D.Q.Appl.Catal. A 2014,469,312.doi:10.1016/j.apcata.2013.10.008
(11) Ding,T.;Qin,Y.N.;Ma,Z.Chin.J.Catal.2002,23(3),227. [丁 彤,秦永宁,马 智.催化学报,2002,23(3),227.]
(12) Wang,F.;Xu,X.L.Chem.Ind.Eng.Prog.2012,31(1),107. [王 丰,徐贤伦.化工进展,2012,31(1),107.]
(13) Wang,R.;Li,C.C.;Chen,T.W.;Lin,J.X.;Mao,S.L.Chin.J. Catal.2004,25(9),711. [王 榕,林墀昌,陈天文,林建新,毛树禄.催化学报,2004,25(9),711.]
(14) Edwards,J.K.;Freakley,S.J.;Carley,A.F.;Kiely,C.J.; Hutchings,G.J.Accounts Chem.Res.2013,47,845.
(15) Hutchings,G.J.;Kiely,C.J.Accounts Chem.Res.2013,46, 1759.doi:10.1021/ar300356m
(16) Sankar,M.;Dimitratos,N.;Miedziak,P.J.;Wells,P.P.;Kiely, C.J.;Hutchings,G.J.Chem.Soc.Rev.2012,41,8099.doi: 10.1039/c2cs35296f
(17) Yang,X.;Du,L.;Liao,S.J.;Li,Y.X.;Song,H.Y.Catal. Commun.2012,17,29.doi:10.1016/j.catcom.2011.10.006
(18) Kittisakmontree,P.;Pongthawornsakun,B.;Yoshida,H.;Fujita, S.;Arai,M.;Panpranot,J.J.Catal.2013,297,155.doi: 10.1016/j.jcat.2012.10.007
(19) Yang,X.;Chen,D.;Liao,S.J.;Song,H.Y.;Li,Y.W.;Fu,Z.Y.; Su,Y.L.J.Catal.2012,291,36.doi:10.1016/j.jcat.2012.04.003
(20) Ouyang,L.;Da,G.J.;Tian,P.F.;Chen,T.Y.;Liang,G.D.;Xu, J.;Han,Y.F.J.Catal.2014,311,129.doi:10.1016/j. jcat.2013.11.008
(21) Menegazzo,F.;Signoretto,M.;Manzoli,M.;Boccuzzi,F.; Cruciani,G.;Pinna,F.;Strukul,G.J.Catal.2009,268, 122.doi:10.1016/j.jcat.2009.09.010
(22) Suo,Z.H.;Ma,C.Y.;Liao,W.P.;Jin,M.S.;Lv,H.Y.Fuel Process.Technol.2011,92 1549.doi:10.1016/j. fuproc.2011.03.018
(23) Pawelec,B.;Venezia,A.M.;La Parola,V.;Cano-Serrano,E.; Campos-Martin,J.M.;Fierro,J.L.G.Appl.Surf.Sci.2005, 242,380.doi:10.1016/j.apsusc.2004.09.004
(24) Wang,Z.Q.;Zhou,Z.M.;Zhang,R.;Li,L.;Cheng,Z.M.Acta Phys.-Chim.Sin.2014,30,2316.[王沾祺,周志明,张 锐,李 莉,程振民.物理化学学报,2014,30,2316.]doi:10.3866/ PKU.WHXB201410152
(25) Edwards,J.K.;Solsona,B.E.;Landon,P.;Carley,A.F.; Herzing,A.;Kiely,C.J.;Hutchings,G.J.J.Catal.2005,236, 69.doi:10.1016/j.jcat.2005.09.015
(26) Bulushev,D.A.;Beloshapkin,S.;Plyusnin,P.E.;Shubin,Y.V.; Bukhtiyarov,V.I.;Korenev,S.V.;Ross,J.R.H.J.Catal.2013, 299,171.doi:10.1016/j.jcat.2012.12.009
(27) Edwards,J.K.;Ntainjua,N.;Carley,A.F.;Herzing,A.A.; Kiely,C.J.;Hutchings,G.J.Angew.Chem.Int.Edit.2009,48, 8512.doi:10.1002/anie.v48:45
(28) Edwards,J.K.;Thomas,A.;Carley,A.F.;Herzing,A.A.;Kiely, C.J.;Hutchings,G.J.Green Chem.2008,10,388.doi:10.1039/ b714553p
(29) Babu,N.S.;Lingaiah,N.;Kumar,J.V.;Prasad,P.S.S.Appl. Catal.A 2009,367,70.doi:10.1016/j.apcata.2009.07.031
(30) Qian,K.;Huang,W.X.Catal.Today 2011,164,320.doi: 10.1016/j.cattod.2010.10.018
(31) Maclennan,A.;Banerjee,A.;Hu,Y.F.;Miller,J.T.;Scott,R.W. J.ACS Catal.2013,3,1411.doi:10.1021/cs400230t
(32) Márquez,A.M.;Graciani,J.;Sanz,J.F.Theor.Chem.Acc. 2010,126,265.doi:10.1007/s00214-009-0703-0
(33) Li,Y.;Feng,J.T.;He,Y.F.;Evans,D.G.;Li,D.Q.J.Ind.Eng. Chem.2012,51,11083.doi:10.1021/ie300385h
(34) Yang,Y.H.;Lin,Y.J.;Feng,J.T.;Evans,D.G.;Li,D.Q.Chin. J.Catal.2006,27(4),304.[杨永辉,林彦军,冯俊婷,Evans, D.G.,李殿卿.催化学报,2006,27(4),304.]
(35) Santacesaria,E.;Di Serio,M.;Velotti,R.;Leone,U.J.Mol. Catal.1994,94,37.doi:10.1016/0304-5102(94)87028-4
Catalytic Performance of PdAu/Al2O3Catalyst with Special Structural and Electronic Properties in the 2-Ethylanthraquinone Hydrogenation Reaction
HAN You HE Zhi-Yuan GUAN Yong-Chuan LI Wei ZHANG Jin-Li*
(School of Chemical Engineering and Technology,Tianjin University,Tianjin 300072,P.R.China)
Aseries of bimetallic PdAu catalysts with different structures were prepared by changing the loading sequence of Pd and Au for the hydrogenation of 2-ethylanthraquinone.Pd/Au/Al2O3was obtained by loading Pd ontoAu particles deposited onto anAl2O3support with a hydrogenation efficiency up to 14.27 g·L-1.According to X-ray diffraction,transmission electron microscopy,hydrogen temperature program reduction,and X-ray photoelectron spectroscopy measurements,the popcorn structure and unique electronic properties of the Pd species in the Pd/Au/Al2O3catalyst resulted in the highest content of surface metallic Pd,which was the most active component for the reaction.What is more,the addition of Au can effectively reduce the amount of degradation products by suppressing side reactions.
Loading sequence;Bimetallic PdAu;Anthraquinone hydrogenation; Degradation product
O643
10.3866/PKU.WHXB201501292www.whxb.pku.edu.cn
Received:November 26,2014;Revised:January 28,2015;Published on Web:January 29,2015.
∗Corresponding author.Email:zhangjinli@tju.edu.cn;Tel:+86-22-27401476.
The project was supported by the National Natural Science Foundation of China(21106094,21276179),National Key Basic Research Program of China(973)(2012CB720300),and Program for Changjiang Scholars,Innovative Research Team in University,China(IRT1161).
国家自然科学基金委(21106094,21276179),国家重点基础研究规划项目(2012CB720300)和长江学者与创新团队发展计划(IRT1161)资助
