微囊藻毒素分子印迹搅拌棒涂层的研制及其吸附性能
2014-12-24丘秀珍郭会时
丘秀珍, 梁 勇, 郭会时*
(1. 韶关学院化学与环境工程学院,广东 韶关512005;2. 华南师范大学化学与环境学院,广东 广州510006)
微囊藻是蓝藻水华中的优势藻,它能产生一类环状七肽结构的微囊藻毒素(microcystins,MC),这类物质已证实与人群中原发性肝癌有很大的关系[1]。微囊藻毒素的种类较多,目前发现的同分异构体大约80 多种,以MC-LR、MC-YR 和MC-RR存在最普遍、含量最多。MC 的毒性和其结构相关,其中MC-LR 是已知毒性最强、危害最大的一种淡水藻类毒素,是最强的肝脏肿瘤促进剂[2]。为了确保饮用水的安全,我国《地表水环境质量标准》(GB 3838-2002)中对生活饮用地表水源地提出微囊藻毒素MC-LR 的限量标准为0.001 mg/L。
目前微囊藻毒素的主要分析方法有超高效液相色谱法[2,3]和液相色谱-质谱联用法[4,5],但这些方法使用之前都需要复杂的样品前处理。分子印迹-搅拌棒吸附萃取技术(molecularly imprinted polymers-stir bar sorptive extraction,MIPs-SBSE)是近年来发展较快的一种新型前处理技术,因具备高选择性、高灵敏度、操作简便和耗溶剂少等优点,已广泛用于痕量有机物的富集,Li 课题组[6-9]对此也作了大量的研究报道。但从目前发展水平来看,分子印迹搅拌棒技术仍存在一些不足,如大部分印迹涂层采用传统聚合方法,搅拌棒涂层形状、厚薄不均,吸附能力较弱,印迹效率不高;MIPs 涂层太厚,严重影响目标化合物的传质速度,在固相萃取应用中,导致模板分子的“泄漏”。这些不足都大大降低了搅拌棒检测的准确度和灵敏度,限制了其应用与发展。
本研究通过可逆加成-断裂链转移(reversible addition fragmentation chain transfer,RAFT)自由基聚合法,在凹凸棒土表面聚合制备了磁性分子印迹聚合物;并将印迹聚合物作为涂层介质,通过溶胶-凝胶法制作搅拌棒,用于富集水体中痕量的微囊藻毒素。在RAFT 反应中,通常加入双硫酯衍生物作为链转移试剂,同时加入其他引发剂,如偶氮二异丁腈。在聚合中链转移试剂与增长链自由基形成休眠的中间体,该中间体可自身裂解,释放出新的活性自由基,结合单体形成增长链,双硫酯衍生物在活性与休眠自由基之间迅速转移,限制了增长链自由基之间的不可逆双基终止副反应,使聚合反应得以有效控制[10]。由于RAFT 聚合具有活性/可控的特点,因此可以通过调整实验条件,控制分子印迹聚合物在基体表面的生长。利用RAFT 自由基聚合法合成分子印迹材料,既可通过合成较厚的分子印迹聚合物膜以获得较高的吸附容量,也可以通过合成较薄的分子印迹聚合物膜以获得较好的吸附效率[10],目前已成为分子印迹合成的研究热点[10-12]。本文采用RAFT 自由基聚合法成功合成了微囊藻毒素分子印迹聚合物,并将其作为搅拌棒涂层成功用于微囊藻毒素的分离富集。
1 实验部分
1.1 仪器与试剂
LC-20AT 高效液相色谱仪,配光电二极管阵列(PDA)检测器(日本岛津公司);DZF-6050D 真空干燥箱(上海齐欣科学仪器有限公司);傅里叶变换红外光谱仪FTIR-8400S (日本岛津公司);透射电子显微镜(TEC,JEOLIEM-200CX,日本)。
微囊藻毒素-LR、微囊藻毒素-RR 标准品(纯度≥98%,Sigma 公司);三氯化铁,氯化亚铁,凹凸棒土(ATP),4-(氯甲基)苯基三氯硅烷,三乙胺,甲苯,苯基溴化镁(1 mol/L,四氢呋喃为溶剂),超干四氢呋喃,二硫化碳,二甲基亚砜,甲基丙烯酸(MAA),乙二醇二甲基丙烯酸酯(EGDNA),偶氮二异丁腈(AIBN),十二烷基硫酸钠(SDS),丙酮,氯仿,乙酸,氨水,以上试剂未注明纯度的均为分析纯;乙腈,甲醇(色谱纯);超纯水。
1.2 磁性分子印迹材料的制备
1.2.1 凹凸棒土磁性复合材料(ATP@Fe3O4)的制备
参照文献[13]采用改进的共沉淀法合成ATP@Fe3O4磁性复合材料。具体步骤如下:称取4.72 g FeCl3·6H2O 和2.1 g 活化的ATP 粉末加入到180 mL 蒸馏水中,超声分散30 min 后,继续搅拌3.0 h,获得稳定的悬浮液。接着在N2保护和剧烈搅拌下加入1.72 g FeCl2·4H2O。随后在80 ℃油浴下逐滴加入10 mL 氨水(25%,质量分数),并持续反应30 min。产物用永磁铁分离,用水清洗后置于真空干燥箱中干燥。
1.2.2 ATP@Fe3O4表面RAFT 链转移剂的制备
制备方法参考文献[12],取1.2 g 干燥的ATP@Fe3O4分散于20 mL 甲苯中,加入1.30 mL 4-(氯甲基)苯基三氯硅烷,6 mL 15% (v/v)的三乙胺甲苯溶液,氮气保护下搅拌反应12 h。反应结束后离心分离沉淀,沉淀用乙醇洗涤多次,60 ℃真空干燥,即得到表面苄基化的磁性凹凸棒土(ATP@ Fe3O4-Cl)。将6 mL 苯基溴化镁分散于10 mL 超干四氢呋喃中,在50 ℃油浴下逐滴加入800 μL 干燥CS2,反应1 h 后加入200 mg ATP@Fe3O4-Cl,氮气保护下于50 ℃油浴反应6 h。产物用磁铁分离,依次用甲醇、丙酮洗涤,60 ℃真空干燥,得到RAFT 链转移剂化的磁性凹凸棒土(ATP@Fe3O4-RAFT)。
1.2.3 凹凸棒土表面引发RAFT 法制备MC-LR分子印迹聚合物
称取30 μg MC-LR 分散于8 mL 二甲基亚砜中,依次加入6 mmol MAA、25 mmol EGDNA、70 mg ATP@ Fe3O4-RAFT、0.05 mg AIBN,分散均匀后通氮除氧10 min,55 ℃油浴反应24 h。反应结束后用磁铁分离沉淀,沉淀依次用氯仿、甲醇-乙酸(9∶1,v/v)洗涤,60 ℃真空干燥过夜。非分子印迹聚合物(NIPs)的制备方法除不加MC-LR 外与分子印迹聚合物的制备方法相同。
1.3 分子印迹搅拌棒涂层的制备
搅拌棒的制作过程参照文献[9],在玻璃棒(30 mm×2.5 mm)内放入一铁芯,两端封死,作为涂渍的载体,玻璃棒在涂渍前依次用NaOH 和HCl 浸泡6 h,用水洗净后置于真空干燥箱内干燥备用。将30 mg MIPs 磁性微球分散在900 μL 的甲苯中,依次加入150 μL 甲基三甲氧基硅烷、450 μL 含氢硅油和900 mg 聚二甲基硅氧烷,充分混匀后,加入240 μL的三氟乙酸,立刻涡旋2 h,直至溶液变为均匀的乳白色胶液。然后,将活化好的玻璃棒插入胶液中,30 min 后将其取出,用氮气吹干后,再次浸没在胶液中,如此反复涂层5 ~6 次。最后,将涂层好的玻璃棒置于真空干燥箱中,室温下老化干燥12 h。依上述操作用NIPs 代替MIPs,制得NIPs 搅拌棒。
1.4 MIPs-SBSE 的萃取性能研究
准确量取5 mL 20 μg/L MC-LR 溶液,置于圆底烧瓶中,分别放入MIPs 和NIPs 搅拌棒,在室温下振荡(200 r/min)吸附。5 h 后,将搅拌棒取出,置于5 mL 甲醇-乙酸(9∶1,v/v)混合溶剂中解吸,解吸液吹干后,加入150 μL 甲醇复溶,溶液经0.22 μm 滤膜过滤后进行HPLC 测定,计算吸附量。以MCRR 为竞争底物,考察MIPs 涂层对MC-LR 的选择性。
1.5 色谱分析条件
色谱柱:Inertstil (ODS-SP)(150 mm × 4.6 mm,5 μm);检测波长:238 nm;流动相:乙腈-水(50∶50,v/v);流速:1.0 mL/min。
2 结果与讨论
2.1 分子印迹聚合物的表征
2.1.1 透射电镜图
ATP@ Fe3O4复合材料和ATP@ Fe3O4-MIPs的透射电镜图见图1,从ATP@Fe3O4-MIPs 电镜图中可以看出,制备的凹凸棒土磁性微球表面有明显的聚合物涂层,这说明其表面已成功聚合MIPs。
2.1.2 红外光谱分析
图2 为ATP@Fe3O4-MIPs 制备过程中各步骤产物的红外光谱图。

图1 (a)ATP@Fe3O4 和(b)ATP@Fe3O4-MIPs的透射电镜照片Fig.1 Transmission electron microscopy (TEM)images of (a)ATP@Fe3O4 and (b)ATP@Fe3O4-MIPs

图2 样品的红外光谱图Fig.2 FTIR spectra for samples
在ATP@Fe3O4的红外光谱图(图2a)中,1 010 cm-1为Si-O-Si 的反对称伸缩振动吸收峰,3 450 cm-1为Si-O-H 的伸缩振动峰。ATP@Fe3O4-Cl 的红外光谱图(图2d)中,1 645 cm-1处出现了苯环骨架-C=C-的伸缩振动吸收峰;910 cm-1处出现了C-Cl 的振动吸收峰。这表明球形硅胶表面的硅羟基和硅烷试剂之间发生缩合反应,将4-(氯甲基)苯基三氯硅烷试剂接枝到ATP 表面。ATP@ Fe3O4-RAFT 的红外光谱图(图2b)中,1 100 cm-1附近-C =S 伸缩振动峰出现,1 645 cm-1处苯环骨架-C =C-伸缩振动吸收峰增强,说明硅胶表面成功负载了RAFT试剂。ATP@Fe3O4-MIPs 微球的红外光谱图(图2c)中,1 700 cm-1附近吸收峰明显增强,归属于功能单体MAA 和交联剂EGDMA 中的-C =O 伸缩振动峰,说明在球形硅胶表面成功合成了MIPs。
2.2 MIPs-SBSE 萃取条件的优化
2.2.1 搅拌棒的稳定性考察
为了考察涂层的热稳定性,我们将MIPs 和NIPs 搅拌棒置于真空干燥箱里,依次在25、40、60℃下加热6 h。结果表明,在这3 种温度下搅拌棒均无明显变化,说明MIPs 和NIPs 在该温度范围内具有良好的热稳定性。此外,为考察涂层的耐溶剂性,我们分别尝试用超纯水、甲醇、乙醇、甲苯浸泡搅拌棒。实验结果显示,MIPs 和NIPs 涂层经纯水、甲醇、乙醇室温下浸泡12 h 后,涂层完好,外观未见有损坏,且吸附试验结果没有明显的改变;而在甲苯中浸泡12 h 后,可见涂层变为透明胶状,且明显发生脱落,这可能是聚二甲基硅氧烷在非极性溶剂中的溶胀作用引起的。这表明MIPs 涂层适合在强极性溶剂中吸附萃取,满足吸附萃取水体中微囊藻毒素的要求。
2.2.2 萃取时间的选择
按1.4 节实验方法,我们考察了萃取时间为1~12 h 时,MIPs 和NIPs 搅拌棒涂层对MC-LR 的吸附量,结果如图3。由图3 可知,MIPs 和NIPs 涂层对MC-LR 的吸附量在0 ~3 h 之内增加较快,3 h后增加较为缓慢,5 h 后达到吸附平衡,且MIPs 涂层吸附量明显高于NIPs。故本实验选择搅拌吸附时间为5 h。
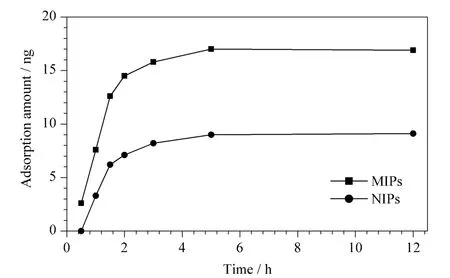
图3 MIPs 和NIPs 涂层搅拌棒的吸附量Fig.3 Extraction amounts of MIPs coated and NIPs coated stir bars
2.2.3 解吸溶剂和解吸时间的选择
实验比较了用甲醇、乙腈、甲醇-乙酸(9∶1,v/v)和乙腈-乙酸(9∶1,v/v)作解吸溶剂,搅拌棒涂层吸附萃取20 mg/L MC-LR 的萃取率,由实验结果可知,甲醇解吸体系的解吸效果稍强于乙腈解吸体系。原因可能是甲醇属于质子型的极性溶剂,其破坏目标物与搅拌棒涂层氢键作用力的能力要强于乙腈分子。在甲醇中加入10% (v/v)乙酸,萃取率略有增加,因此,本研究选择甲醇-乙酸(9∶1,v/v)作为解吸溶剂。实验同时考察了解吸时间为10 ~90 min 时,MIPs 和NIPs 涂层对MC-LR 的萃取率。结果表明解吸时间在30 min 前,萃取率逐渐增大,30 min 后萃取率变化趋于平缓。说明涂层吸附的MCLR 在30 min 基本解吸完全。故选择30 min 为解吸时间。
2.3 MIPs-SBSE 的选择性能研究
在自然界含量最多的3 种MC 同分异构体中,MC-LR 的急性毒性最强,MC-YR 次之,MC-RR最弱。目前,国内外研究最多的主要是MC-LR 和MC-RR,故本研究也以这两种物质进行竞争选择性试验。配制相同浓度的MC-LR 与MC-RR 标准溶液。在最佳萃取条件下,分别用MIPs-SBSE 和NIPs-SBSE 萃取,计算回收率(见图4)。由实验结果可知,MIPs 搅拌棒对MC 的回收率明显高于NIPs 搅拌棒,且MIPs 搅拌棒对MC-LR 的萃取回收率高于MC-RR,说明MIPs 搅拌棒对MC 具有良好的选择性,对模板分子MC-LR 选择性更佳。
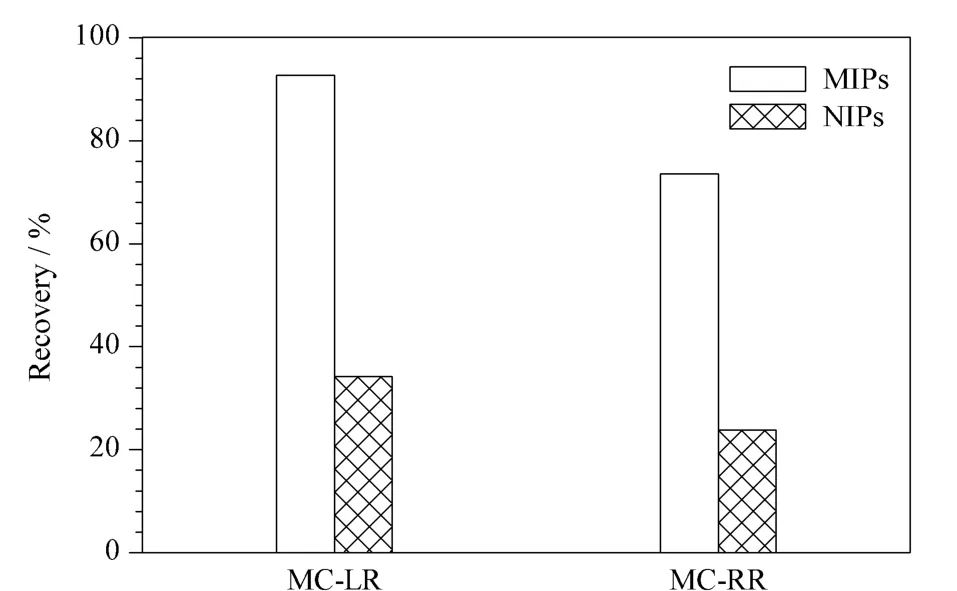
图4 MIPs 和NIPs 搅拌棒对MC-LR 的选择性吸附Fig.4 Selective adsorption of MC-LR MIPs and NIPs coated stir bars
2.4 搅拌棒重复利用性能研究
取饱和吸附MC-LR 的MIPs 搅拌棒,以5 mL甲醇和乙酸的混合溶液(90∶10,v/v)为解吸溶剂,搅拌解吸30 min,使MC-LR 从MIPs 涂层上洗脱下来。再按以上方法进行5 次吸附-解吸附过程。计算MIPs 搅拌棒涂层对MC-LR 5 次再生的吸附容量。研究发现,5 次再生后,MIPs 搅拌棒涂层对MC-LR 的吸附容量仅降低了5.7%,表明搅拌棒涂层具有良好的重复利用性能。
2.5 方法的可行性和实际应用
我们将MIPs 搅拌棒吸附萃取与HPLC-PDA 联用,用外标法测定了饮用水体中的MC-LR,该方法在0.010 ~5.0 mg/L 范围内线性关系良好,r2>0.997,检出限为0.27 μg/L (S/N =3)。我们用该方法测定了广东韶关居民饮用水源区水体中的微囊藻毒素MC-LR。直接用饮用水进样,色谱图中检测不到MC 的色谱峰;饮用水经MIPs-SBSE 富集后进样,MC-LR 和MC-RR 可以被检出(见图5),富集倍数分别为17.6 和8.2,说明MIPs-SBSE 对MC具有较好的富集效果。同时我们还做了样品加标试验,饮用水中MC-LR 添加水平为20、40、80 μg/L时,加标回收率范围为83.33% ~100.07%,且相对标准偏差(RSD,n =3)为1.40% ~9.17%(见表1),结果满足痕量分析的要求。
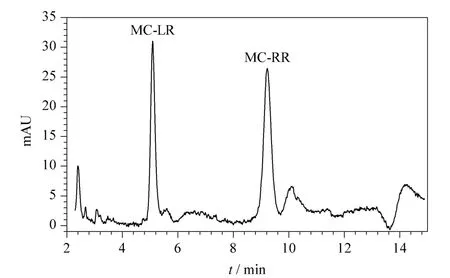
图5 富集后实际样品的色谱图Fig.5 Chromatogram of a real sample after enrichment

表1 水样加标回收率与精密度(n =3)Table 1 Recoveries and RSDs of the analytes spiked in water samples (n =3)
3 结论
我们采用RAFT 聚合法利用ATP@Fe3O4制备了MC-LR 磁性分子印迹微球,并通过溶胶-凝胶法,将合成的印迹微球分散在合成的乳白色胶液中,将搅拌棒插入其中制得分子印迹搅拌棒。在优化的萃取条件下,分子印迹搅拌棒显示出了较好的选择吸附性能,MIPs 搅拌棒对MC 的吸附能力也明显优于NIPs 搅拌棒。同时我们建立了MIPs-SBSEHPLC 联用检测饮用水中痕量微囊藻毒素的新方法,该方法的富集倍数高,方法检出限低至0.27 μg/L,显示了较高的灵敏度。最后,我们应用该方法成功检测了水源区水体中的MC-LR,结果令人满意。
致谢:感谢韶关学院化学与环境工程学院卢文贯教授为本论文的测试工作提供的帮助。
[1] Shen Q,Cui L F,Zhao S,et al. Chinese Journal of Analytical Chemistry (申晴,崔莉凤,赵硕,等. 分析化学),2012,40(3):442
[2] Zhang M,Tang F L,Chen F. Chinese Journal of Chromatography (张明,唐访良,陈峰,等. 色谱),2012,30(1):51
[3] Li B,Liu W,Fan S,et al. Chinese Journal of Chromatography(李兵,刘伟,范赛,等. 色谱),2012,30(6):584
[4] Guo J,Yang X L,Ye M L. Chinese Journal of Analytical Chemistry (郭坚,杨新磊,叶明立. 分析化学),2011,39(8):1256
[5] Shi J,Jing S M,Lu F,et al. Chinese Journal of Chromatography(施俭,景澍闽,陆峰,等. 净水技术),2013,32(3):52
[6] Hu Y L,Li J W,Hu Y F,et al. Talanta,2010,82:464
[7] Xu Z G,Song C Y,Hu Y L,et al. Talanta,2011,85:97
[8] Huang J X,Hu Y F,Hu Y L,et al. Talanta,2011,83:1721
[9] Xu Z G,Hu Y F,Hu Y L,et al. J Chromatogr A,2010,1217:3612
[10] Zhang Y,Dong X C. Journal of Instrumental Analysis (张毅,董襄朝. 分析测试学报),2008,27(10):1025
[11] Dai J D,Pan J M,Xu L C,et al. J Hazard Mate,2012,205/206:179
[12] Zhou W H,Lu C H,Yin X F,et al. Journal of Fuzhou University:Natural Science Edition (周文辉,卢春华,尹晓斐,等. 福州大学学报:自然科学版),2008,36(2):284
[13] Pan J M,Xu L C,Dai J D,et al. Chem Eng J,2011,174(1):68
