人源甲型H1N1流感HA基因进化特征分析
2014-07-09金梦哲綦朝晖李素丽
金梦哲,綦朝晖,李素丽
(石家庄铁道大学 信息科学与技术学院,河北 石家庄 050043)
过去的三次流感大爆发(1918年由H1N1引发的西班牙流感,1957年由H2N2病毒引发的亚洲流感和1968年由H3N2引发的香港流感)都并非起源于人类,源头实际为禽流感病毒的变异[1]。最近一次甲型H1N1流感疫情在2009年4月大规模爆发于墨西哥,研究人员从墨西哥和美国的感染人群中收集到的流感病毒未曾有过记录。在短短不到一个月的时间里,四十多个国家证实了甲型H1N1流感的大规模爆发[2]。2009年6月11日,WHO宣布将甲型H1N1流感大流行警告级别提升为6级,全球进入流感大流行阶段,H1N1流感已席卷全球160多个国家和地区。直到2010年,WTO宣布甲型H1N1流感的大流行已结束,但据统计目前流感高峰期的主流病毒仍为甲型H1N1流感病毒[3]。2013年底H1N1流感再度流行,美国联邦疾病防治中心12月27日宣布,流感目前已经扩散到全美十个州,引起四名儿童死亡,上万成年人入院治疗。所幸的是,这一病毒尚未发生大的抗原性变异。
随着生物信息学在生命科学的研究中的兴起,使得以计算机为工具对生物信息进行分析和计算的方法成为生物特征信息获取的重要途径。利用生物信息学方法对生物序列进行分析已成为人们的研究热点。在目前对高维度空间中的生物序列的研究中,数据降维方法被成功地用来获取低维数据以便进一步分析。利用生物信息学方法对病毒序列进行表达、分析病毒的进化特征对于预防流感的大爆发有着重要作用。
1 甲型H1N1病毒的HA基因样本序列
流感大爆发与流感病毒编码蛋白中的血凝素(HA)基因的变异密切相关,对于流感病毒的变异和传播而言,最关键的是HA抗原所引起的人体免疫应答[4],因此本文选择以HA基因样本序列来研究H1N1流感病毒的变异规律。
1.1 病毒样本
本文所应用的数据集为从美国国家生物技术信息中心(NCBI)的流感病毒资源库(Influenza Virus Resource,http://www.ncbi.nlm.nih.gov/genomes/FLU/Database/nph-select.cgi?go=database)获取的H1N1病毒HA基因序列源数据。该流感病毒数据库开放性地提供了流感宿主,感染国家、地区,蛋白质类型,病毒亚型,序列长度和收集时间为分类的流感病毒的核苷酸序列和氨基酸序列数据。考虑到2009年4月开始的甲型H1N1流感大爆发,具体的索引数据集设定为2009年3月到8月的长度不限的人源甲型H1N1流感病毒HA基因的蛋白质全序列,如图1所示。
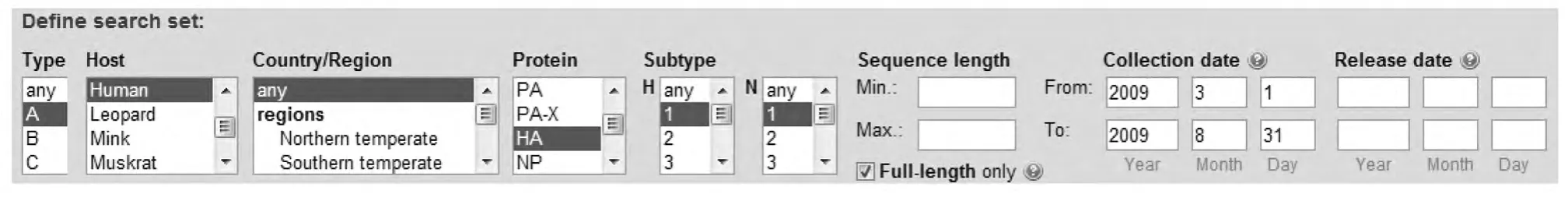
图1 索引收集时间为2009年3月1日到8月31日的H1N1病毒HA基因序列
满足条件的甲型H1N1流感病毒HA基因序列共有3602条,按数据保存于FASTA文件proteinHA.fas中。
1.2 MUSCLE对位
图2是proteinHA.fas中三条流感病毒原始序列的数据形式。每条H1N1流感病毒HA基因序列由两行数据组成:第一行为该序列的描述,依次为该病毒HA序列的GenBank索引号,收集地点和时间等;第二行为蛋白质序列的氨基酸排列。经过查询后,满足条件的3602条序列有长有短,序列的长短不同在序列的聚类分析过程中造成很大的干扰。为了使聚类分析不受序列长度的影响,采用经典的蛋白质水平上多序列比对 MUSCLE(Multiple Protein Sequence Alignment)[5]方法。在算法层面上,MUSCLE是一个渐进比对并结合迭代优化的综合算法,该对位不但要求对位后序列长度均相等,而且不同序列在相同位置尽可能多地存在相同片段[6]。

图2 H1N1流感病毒序列的部分原始数据

图3 H1N1流感病毒序列MUSCLE对位后的部分数据
MUSCLE对位排列后的流感病毒序列如图3所示,对某些序列氨基酸不足的位置用“-”占位,这样造成了序列的错位使得在相同位置更多的同种氨基酸片段得以产生。MUSCLE对位后,该流感病毒序列样本中3602条甲型H1N1流感病毒HA基因序列长度均为582。
2 基于PCA的聚类分析
经过MUSCLE对位排列后的流感病毒序列,本身具有对位病毒序列的相同位置越多相似程度越高的特征。一次流感大爆发简单地说是由流感病毒结构发生的变异引起的,对于大爆发期间获取到的数据量相当大的流感病毒样本来说,其中既有大爆发前通过变异产生的新型病毒菌株,又有旧的常年随季节变化的病毒菌株。在研究流感大爆发的传播特征时旧病毒是没有产生作用的,因此需要挑选新型病毒菌株,这就产生了对H1N1病毒数据样本中的序列的进行分类的思考。
2.1 病毒序列的数字化表达
MUSCLE对位后的序列本身有等长的性质,但是要进行对位后的序列数字化聚类分析,首先应该对病毒的HA基因样本序列数字化表达,使其转化为数值序列。
构成蛋白质的氨基酸一共有二十种,简写分别为:P,L,Q,H,R,S,F,Y,W,C,T,I,M,K,N,A,V,D,E,G。为了能够使得序列中所有的氨基酸排列信息都保存在数值序列中,必须要求病毒序列的数字化表达后形成的数值序列与原氨基酸字母序列能够形成一一映射关系。本文引入Xiao等人氨基酸编码的二进制符号语言的方法[7],将氨基酸符号序列通过二进制编码数字化表达为数值序列,具体氨基酸字母对应的编码如表1所示。
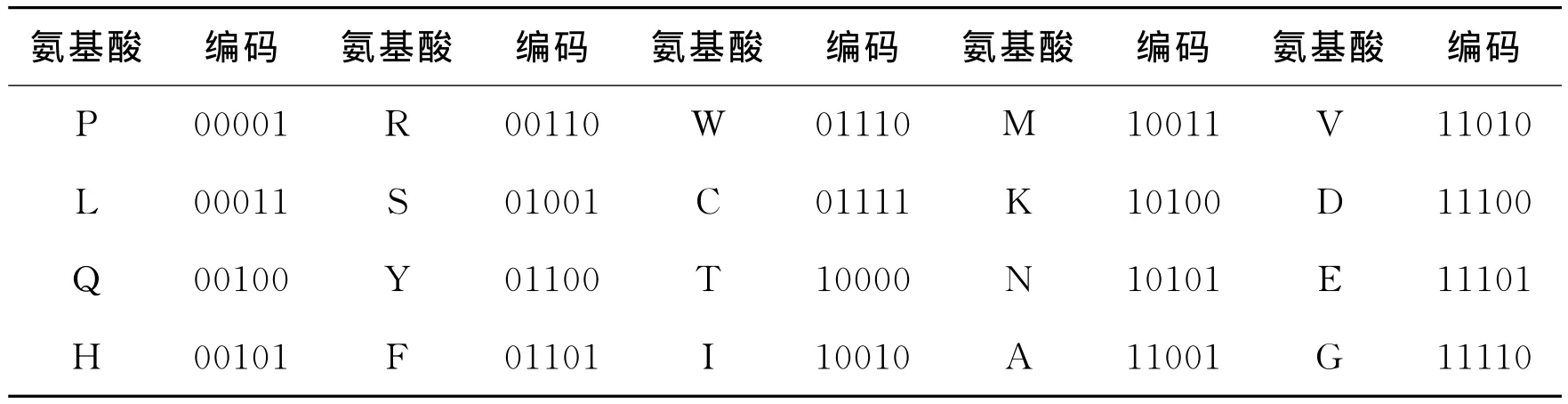
表1 二十种氨基酸的二进制编码
文献[7]中数字化表达病毒序列的方法,是通过相似原则、互补原则和分子识别理论制定的,可以完整地捕捉到氨基酸序列的物理和化学特性。此外,由于1.2节MUSCLE对位后的序列中会产生“-”占位符,本文在文献[7]数字表达序列的方法基础之上利用00000表示“-”。在原始序列中会有一些位置由于统计失误等原因产生其他字符,这些字符实际上没有意义,统一编码为00000。原始序列中还会使用“X”代表任意氨基酸,考虑到任意氨基酸位与占位符、无意义位的严格区别,我们将“X”编码为11111。
这样,之前得到的MUSCLE对位后的HA基因序列样本就表达成由0、1组成的数值序列样本。由于采用了5位二进制编码,数值序列的长为对位后字母序列长度的5倍,即数值序列长度为2910。
2.2 PCA降维方法
主成分分析(Principal Component Analysis,PCA)从统计学的角度来说是一种多元统计方法,它可以将多个变量通过线性变换选出较少的重要变量。具体地说,PCA通过线性变换求出原数据矩阵到主元得分空间的映射矩阵,选取最重要的部分,将其余的维数省去,达到降维的目的[8]。PCA方法间接地对数据进行了压缩处理,同时很大程度上保留了原数据的信息。
经过2.1节病毒序列的数字化表达后,3602条甲型H1N1的HA蛋白序列数据转化为3602×2910的高维数值矩阵,样本数量为3602,每个样本有2910个特征点。将矩阵简化记为:
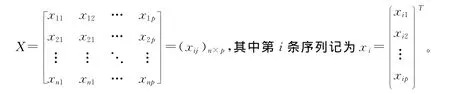
MATLAB提供了所需的主成分分析,只需在软件中调用PCA命令princomp,即可实现对数字化表达后的高维数据的降维,具体的调用格式如下:
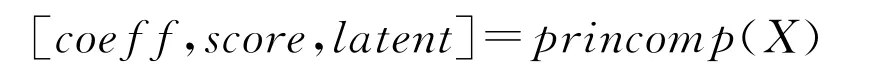
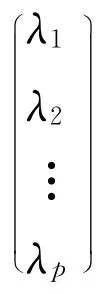
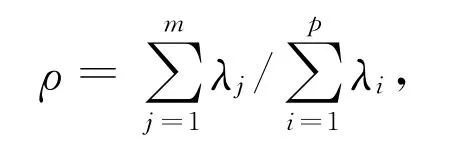
其中m为选取的score矩阵的列数,也就是选取的维数。随着m的增大,选取的score矩阵的列数增加,ρ也逐渐趋近于1。为了保证选取的主成分能够忽略掉不重要的成分,同时不会丢失太多的重要成分,这里要求ρ≥0.85[9]。
通过计算,m=1时,ρ=0.8322,m=2时,ρ=0.8769≥0.85,于是选取score的前两列作为原矩阵X的主成分进行提取,得到一个n×2的降维矩阵。这样,甲型H1N1流感病毒HA蛋白全序列数据集组成的3602×2910的原数值矩阵通过主成分分析降维方法得到了3602×2的数值矩阵。通过PCA降维方法,高维数值矩阵在保存了其数值特性的前提下降维成二维数值矩阵,即一条序列可以简单地用两个主成分得分来表示。
2.3 病毒样本分类
将数值序列经PCA得到的第一主成分作为x轴坐标,第二主成分作为y轴坐标,即可将3602条甲型H1N1流感病毒HA基因序列样本表达在平面上,如图4(a)所示。
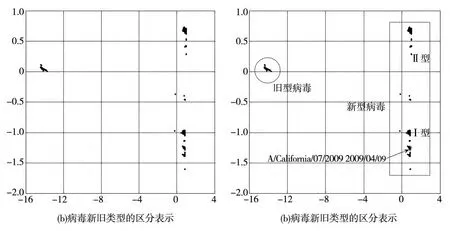
图4 病毒样本的二维图像表达
由于PCA中score矩阵是原数值矩阵通过线性变换得到的,因此图4(a)上的点和原病毒序列之间存在着映射的关系。图4(a)中,虽然3602条病毒序列样本均属于甲型流感病毒H1N1亚型,但是根据主元分析后的第一主成分得分,这些序列大致可以划分成了两个特征显著不同的病毒簇:第一主成分在-14附近的病毒簇和第一主成分在0到1的病毒簇。
3 H1N1流感HA基因进化特征分析(2009.3-2009.8)
3.1 病毒的新旧区分
在2009年的甲型H1N1流感大爆发期间采集到的病毒菌株中,既有流感大爆发之前许多年里一直存在的没有高致病性的旧病毒,也有在大爆发前或者大爆发期间产生的新变异的高传染性的流感病毒。对病毒的新旧区分能够对研究病毒的进化特征找到正确的靶标,使得研究结果更加准确。
利用PCA方法对2009年3月到8月采集到的3602个病毒数据样本进行了降维,并对降维得到的低维数据进行了两类病毒簇的分类。经过分类后的病毒序列的氨基酸构成相似,因而具有相近的生物性质。但是,到目前为止还无法确定图4(a)中哪一区域是新型病毒簇,哪一区域是旧型病毒簇。
世界卫生组织推荐以A/California/07/2009 2009/04/09作为甲型H1N1流感疫苗的参考菌株[10],它代表着2009年流感大爆发中的新型病毒群体。因此,A/California/07/2009 2009/04/09病毒菌株一定属于新型流感病毒簇。图4(b)中,右下角的“*”号位置标示出了该病毒样本在二维平面图中的位置,坐标为(0.8556,-1.2513)。在图像上即可判断出矩形区域表示新型流感病毒簇,圆形区域对应为旧流感病毒簇。除此之外,可以看出圆形区域集中在一个相对小的范围内,说明旧病毒的变异程度很低,然而矩形区域的范围相对来说较大,这就代表了新型流感病毒的变异程度高,发生了频繁的抗原飘移。
3.2 病毒传播分析
通过PCA降维方法,原本高维的病毒序列样本转换成了二维数据,该样本数据在平面上可以清晰地表现出来。我们将病毒样本按菌株的收集时间顺序排序,把排序序号作为z轴,即可得出流感病毒数据集的三维表示,如图5:
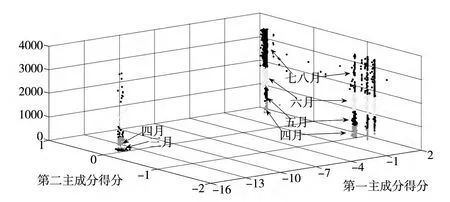
图5 病毒样本的三维图像表达
图5显示了第一、二主成分在时间序列上的变化,箭头指向了在3月到8月收集到的H1N1流感病毒菌株群。通过观察,所有3月份收集到的菌株均属于旧型病毒簇,正与3月流感大爆发尚未发生这一事实相符。而4月开始,流感大爆发于墨西哥,新型H1N1病毒开始广泛传染,也与4月收集到的流感菌株一部分来自旧型病毒簇一部分来自新型病毒簇的事实相符。5、6、7、8四个月流感大爆发,收集到的流感菌株数量很大,且绝大部分属于新型病毒簇。图4(b)中,新型变异病毒由于抗原漂移造成的小幅度变异也分为几个簇,但由于差异很小,只体现在了第二主成分的差异。在图5中也可以看到,从4月开始数据样本的第二主成分较多地在-1附近,而随着时间推移到8月,数据样本的第二主成分更多地分布在0.5到1,说明这些新型病毒随着时间也在发生着细微的变化,在图4(b)中,逐渐由I区向II区变化,但是这些微小变异并未造成抗原转变而形成新的亚型。
4 结论
以2009年3月到8月的3602条甲型H1N1病毒HA基因全序列为数据样本,将经MUSCLE比对的符号序列数字化表达成一组等长的高维数值序列。而后以高维数值序列为数据矩阵,利用主成分分析(PCA)映射到得分空间,选取其中两个主要成分进行降维,得到了病毒样本的二维平面表达图,利用H1N1流感疫苗制作的参考菌株在图中确定并区分了新旧病毒类型,其中新型病毒对于研究流感大爆发中病毒的传播有关键性作用。添加收集时间为第三维度,得到三维图,通过第一、二主成分随时间的变化分析了甲型H1N1流感病毒随时间的变化,得到了甲型H1N1流感病毒HA基因在2009年3月到8月期间的进化特征。
[1]GARTEN R J,DAVIS C T,RUSSELL C A,et al.Antigenic and genetic characteristics of swine-origin 2009A (H1N1)influenza viruses circulating in humans[J].Science,2009,325(5937):197-201.
[2]Malik Peiris J S,POON L L M,GUAN Y.Emergence of a novel swine-origin influenza A virus(S-OIV)H1N1virus in humans[J].Journal of Clinical Virology,2009,45(3):169-173.
[3]GUNSON R N,CARMAN W F.During the summer 2009outbreak of"swine flu"in Scotland what respiratory pathogens were diagnosed as H1N1/2009?[J].BMC infectious diseases,2011,11(1):192.
[4]李国强.流感病毒H1亚型血凝素单抗库的构建及其抗原性变异分析[D].福建:厦门大学,2009.
[5]EDGAR R C.MUSCLE:multiple sequence alignment with high accuracy and high throughput[J].Nucleic acids research,2004,32(5):1792-1797.
[6]杨凡.生物序列分析中若干问题的研究[D].电子科技大学,2011.
[7]XIAO X,SHAO S,DING Y,et al.Using cellular automata to generate image representation for biological sequences[J].Amino Acids,2005,28(1):29-35.
[8]JACKSON J E.A user's guide to principal components[M].John Wiley & Sons,2005.
[9]QI Zhao Hui,WEI Ruo Yan.A combination dimensionality reduction approach to codon position patterns of eubacteria based on their complete genomes[J].Journal of theoretical biology,2011,272(1):26-34.
[10]World Health Organization.Recommended viruses for influenza vaccines for use in the 2010-2011northern hemisphere influenza season[J].Wkly Epidemiol Rec,2010,85(10):81-92.
