8-氧-2'-去氧鸟嘌呤核苷水解机理的理论研究
2014-06-06梁晓琴
郑 妍,尤 勇,梁晓琴
(四川师范大学化学与材料科学学院,成都610066)
1 引 言
基因信息由DNA链上的碱基确定,这些碱基的任何改变都将大大的影响遗传物质的整体性.然而这些碱基可能被氧化损伤,这种氧化是由活性氧(reactive oxygen species,ROS)引起的一种化学修饰,不仅电离辐射或者长波紫外线(UVA)照射可产生活性氧,而且由尼古丁和烟草烟雾引起的有机自由基也可产生活性氧.另外,正常细胞的新陈代谢也能引起碱基的损伤.
DNA中糖苷键的断裂是一个重要的过程,在自然界中发生的频率非常高.然而,DNA的稳定性使得糖苷键断裂成为一个高能量的反应,需要各种酶的催化[1].例如,参与DNA修复过程的DNA糖苷酶使糖苷键断裂,从而将受损的核苷碱基从双螺旋结构中切除.已经提出的不同酶催化DNA糖苷键断裂的机理具有许多相似之处.特别的是由于核苷的稳定性,所提出的机理包括一个亲核试剂的进攻(例如,在水解酶、蓖麻蛋白毒素A链和一些DNA糖苷酶中的水分子、一些DNA糖苷酶的一个活性部位氨基酸和磷酸酶的磷酸氢盐)[1-6].有些酶催化糖苷键断裂的机理包括类氧碳正离子过渡态的形成.例如,已提出的DNA糖苷酶(尿嘧啶DNA糖苷酶,UDG)水解机理包括核苷碱基阴离子的形成,该阴离子的产生是由于碱基-核糖键的断裂,并且假设酶通过其活性部位残基与碱基阴离子中间体之间的氢键相互作用来稳定碱基阴离子中间体.而且,尽管一些糖苷酶(MutY)被认为在碱基离去之前质子化嘌呤或嘧啶,但是有证据表明其他一些受损嘌呤(8-氧鸟嘌呤)能通过阴离子中间体的形成来修复受损核苷[7].另外,机理包括或至少部分包括离去核苷碱基的稳定,这种稳定是通过离去核苷碱基与酶的活性位点氨基酸之间氢键相互作用来达到的.
计算化学能提供有关短期存在的过渡态的有用信息,这种过渡态在实验中很难得到.事实上,很多研究运用计算化学方法来研究糖苷键断裂的路径.然而,这些研究通常仅仅集中在糖苷键被特定酶催化断裂.由于这些酶催化反应机理的相似性,我们认为在不考虑核苷碱基在特定活性部位的相互作用的情况下,研究这种反应,即研究酶催化糖苷键断裂的基本原理是必要的.为了更好的了解核苷碱基在糖苷键裂解过程的活化作用,Wetmore[8]等人作了大量的工作.他们研究了氢原子结合到正常的核苷碱基或者是受损的核苷碱基对碱基酸性的影响以及尿嘧啶核苷的N-糖苷键的水解机理.在目前的工作中,用计算化学的方法研究了有单个水分子参与的8-氧-2'-去氧鸟嘌呤核苷的N-糖苷键水解反应机理.选择水分子作为亲核试剂不仅是由于水在生物体中的重要性,而且还由于水在由几种酶催化的糖苷键断裂过程中的重要作用.
2 计算原理和方法
为了减小去氧核糖糖环上羟基与亲核试剂水分子和碱基的环外O8原子之间的氢键相互作用,在本文最终使用的计算模型中,用甲氧基代替了糖环上的O5'-H羟基基团和O3'-H羟基基团.
气态中,用密度泛函B3LYP方法在标准的6-31++G(d,p)基组对8-oxodG的水解反应进行了研究[9].对各反应途径的反应物、过渡态及产物的几何构型进行了优化,得到各驻点的能量,在优化的几何构型基础上进行了振动频率计算,对各优化驻点的总电子能量进行了零点振动能校正,并从过渡态分别向产物方向和反应物方向进行了内禀反应坐标(IRC[10])计算.所有的过渡态都通过振动分析和IRC计算而得以确认.非限制自旋UB3LYP方法用于开壳层体系的解析计算,稳定点的性质通过在相同水平上的振动频率分析来确认:每个稳定点的所有频率为正,过渡态通过振动模式分析,只含有一个虚频,并且虚频的振动方向沿着反应坐标方向.在298.15K和一个标准大气压下通过频率分析我们也得到零点能(ZPVE)、热量校正、焓和吉布斯自由能.
在相同水平下,应用自然键轨道(NBO)[11]计算了自然原子电荷和键级,并以气态优化结构,考虑水作为溶剂,用极化连续模型[12,13]溶剂效应.所有的计算用Gaussian03程序包计算[14]
3 结果与讨论
3.1 各稳定点的几何结构
对于8-oxodG与单个水分子作用的水解反应中,一方面,我们考虑水分子有三种进攻方式,即水分子可以从去氧核糖糖环的前方、下方和上方进攻异头碳C1'(图2);另一方面,8-氧鸟嘌呤碱基的O8原子和N9原子都可以摘取去氧糖糖环的Ha2'.
当水分子按i)方向进攻时,去氧核糖的环内键O4'-C1'键断裂,从而使糖环开环.因为糖环开环是双功能DNA糖苷酶催化N-糖苷键断裂的步骤之一.这些酶用其一个氮端的氨基基团作为亲核试剂,先打开糖环再与开环产物形成一个席夫碱,从而得到最终产物.然而,在我们的理论模型中,亲核试剂不是氨基基团,而是水分子,因此不可能形成席夫碱.而且,计算结果表明,水分子以i)方向亲核进攻得到的开环中间体能量非常高(约60kcal/mol),因此该反应机理在后文中不予考虑,只考虑另外两种进攻方式.事实上,Zoltewicz[15]和他的的合作者的实验研究表明,一些嘌呤核苷的酸催化水解包括了C-O键断裂,水解机理为:第一步是核苷碱基质子化,第二步是N-糖苷键断裂.
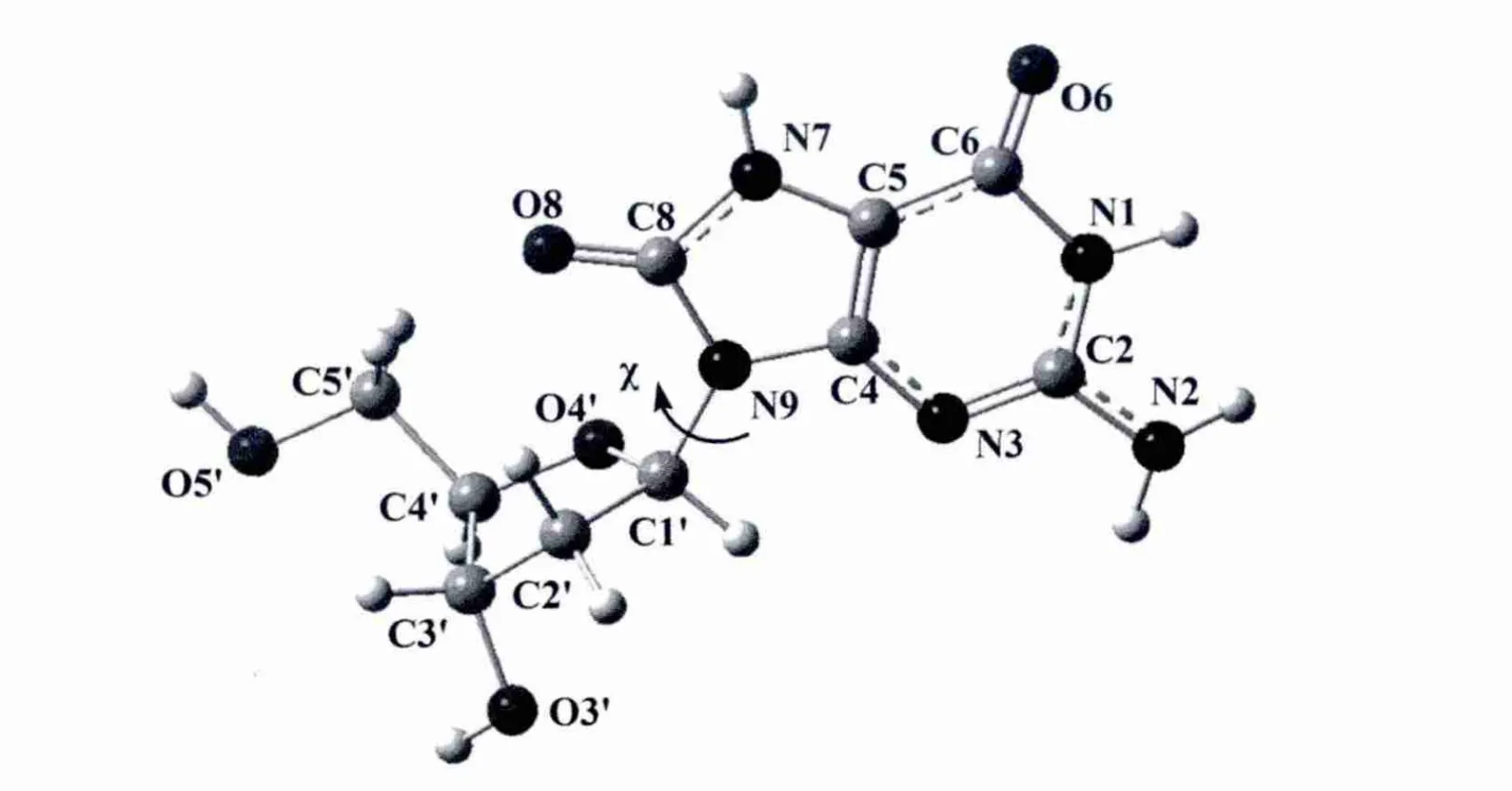
图1 8-氧-2′-去氧鸟嘌呤核苷的结构和原子编号Fig.1 Structures of 8-oxo-2'-deoxyguanosine and atomic numbers
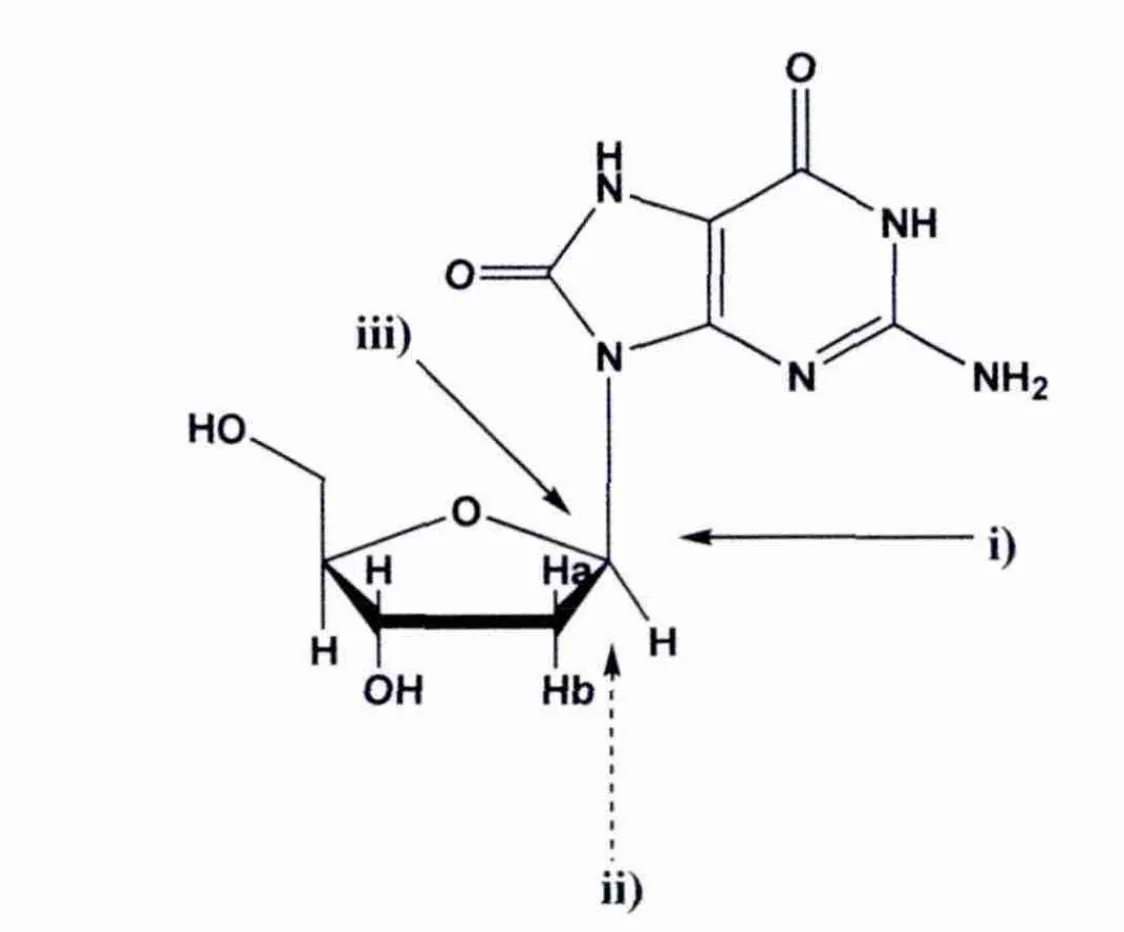
图2 水分子进攻8-氧-7,8-二氢-2′-去氧鸟嘌呤核苷的三种进攻方式Fig.2 Three ways of 8-oxo-2'-deoxyguanosine attacked by water
因此,在本研究工作中,我们考虑了四条反应通道,即ii)进攻方向上的O8摘氢反应机理和N9摘氢反应机理,以及iii)进攻方向上的O8摘氢反应机理和N9摘氢反应机理.在去氧核苷的水解反应中,最重要的步骤是N-糖苷键的断裂,因此,我们将主要关注N-糖苷键的断裂的能量和结构,即反应第一步的能量和结构.
途径一 在第一条反应途径中,亲核试剂水分子从去氧核糖糖环的下方进攻异头碳C1'原子并且由8-氧鸟嘌呤的N9原子摘取去氧糖糖环的Ha2',这条反应途径意味着异头碳C1'的结构反转.如图3所示,在这种情况下,反应的第一步通过将去氧核糖糖环的Ha2'质子转移到碱基的N9原子产生一个类二氢呋喃中间体.第二步包括水分子加成到C-C双键上,而势能剖面图和反应物、过渡态、中间体和产物的结构见图3.
水分子先与受损的去氧鸟苷作用形成8-氧去氧鸟苷-水复合物Ra,我们把Ra作为该反应通道的反应物.紧接着N-糖苷键发生扭转,使二面角O4'-C1'-N9-C4逐渐减小,在反应物中为261.46°,减小到第一个过渡态N-TSa1中的229.66°,但仍然保持反式结构;同时N-糖苷键逐渐拉长,由反应物中的1.455到N-TSa1中的2.965,从图中可看出,N-TSa1结构的N9-C1'键已经断裂,且键级为零.而Ha2'氢原子与N9原子的距离也逐渐缩短,直到 N-TSa1中的1.879Å.而从 NTSa1到第一个中间体N-INTa1的过程中,N9原子已与Ha2'质子相结合.而质子化的受损碱基发生翻转,不仅使二面角 O4'-C1'-N9-C4逐渐增大,由 N-TSa1中的229.66°,增加到 N-INTa1的254.32°,但仍然保持反式结构;而且使碱基环外氨基的Ha2与OW之间发生氢键相互作用,OW与碱基上Ha2的距离为1.866Å,键角OW-Ha2-N2为172.74°.

图3 以途径一进行的8-氧-2′-去氧鸟嘌呤核苷水解吉布斯自由能剖面图(kcal/mol)和优化结构及部分结构参数(键长:Å)Fig.3 Gibbs free energy cross-section(kcal/mol)of 8-oxo-2'-deoxyguanosine hydrolysis according to the first pathway and optimized structures along with their selected configuration parameters(bond lengths(inÅ)).
途径二 在第二条反应途径中,水分子从糖环的上方进攻异头碳C1'原子并且由8-氧鸟嘌呤的N9原子摘取去氧糖糖环的Ha2',这条反应途径意味着异头碳C1'的结构保持.从图4中可以看出,在这种情况下,反应的第一步通过将去氧核糖糖环的Ha2'质子转移到碱基的N9原子产生一个类二氢呋喃中间体.第二步包括水分子加成到C-C双键上,而势能剖面图和反应物、过渡态、中间体和产物的结构见图4.
水分子先与受损的去氧鸟苷作用形成8-氧去氧鸟苷-水复合物Rb,我们把Rb作为该反应通道的反应物.紧接着N-糖苷键发生扭转,使二面角O4'-C1'-N9-C4逐渐减小,在反应物中为257.81°,减小到第一个过渡态N-TSb1中的229.30°,但仍然保持反式结构;同时N-糖苷键逐渐拉长,由反应物中的1.456Å到N-TSb1中的3.034Å,从图中可看出,N-TSb1结构的N9-C1'键已经断裂,且键级为零.而 Ha2'氢原子与OW间的距离以及HaW原子与N9原子的距离也逐渐缩短,分别由反应物中的2.626Å和3.808Å缩短到N-TSb1中的1.427Å和1.398Å,键角 OW-HaW-N9和C2'-Ha2'-OW分别为173.10°和171.06°.而从 N-TSb1到第一个中间体N-INTb1的过程中,N9原子与HaW质子相结合,而OW与Ha2'质子相结合.二面角 O4'-C1'-N9-C4逐渐增大,由 N-TSb1中的229.30°,增加到N-INTb1的253.39°,但仍然保持反式结构;碱基环与新形成的水分子之间发生氢键相互作用,OW与碱基N9上的HaW(由亲核试剂水分子转移到碱基上的质子)的距离为1.875Å,键角 OW-HaW-N9为174.30°.
途径三 在第三条反应途径中,亲核试剂水分子从去氧核糖糖环的下方进攻异头碳C1'原子并且由8-氧鸟嘌呤的O8原子摘取去氧糖糖环的Ha2',这条反应途径意味着异头碳C1'的结构反转.从图5中可以看出,在这种情况下,反应的第一步通过将去氧核糖糖环的Ha2'质子转移到碱基的O8原子产生一个类二氢呋喃中间体.第二步包括水分子加成到C-C双键上,而势能剖面图和反应物、过渡态、中间体和产物的结构见图5.
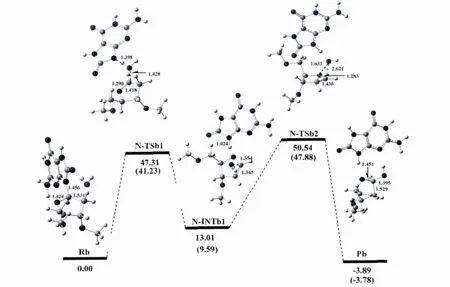
图4 以途径二进行的8-氧-2′-去氧鸟嘌呤核苷水解吉布斯自由能剖面图(kcal/mol)和优化结构及部分结构参数(键长:Å)Fig.4 Gibbs free energy cross-section(kcal/ mol)of 8-oxo-2'-deoxyguanosine hydrolysis according to the second pathway and optimized structures along with their selected configuration parameters(bond lengths(inÅ)).
水分子先与受损的去氧鸟苷作用形成8-氧去氧鸟苷-水复合物Ra,我们把Ra作为该反应通道的反应物.紧接着N-糖苷键发生扭转,使二面角O4'-C1'-N9-C4逐渐增加,在反应物中为261.46°,增加到第一个过渡态中O-TSa1的263.36°,但仍然保持反式结构;同时N-糖苷键逐渐拉长,由反应物中的1.468Å到O-TSa1中的2.644Å,从图中可看出,O-TSa1结构的N9-C1'键已经断裂,且键级为零.而Ha2'氢原子与O8原子的距离也逐渐缩短,直到 O-TSa1中的1.395Å.而从 OTSa1到第一个中间体O-INTa1的过程中,O8原子已与 Ha2'质子相结合.二面角 O4'-C1'-N9-C4继续逐渐增大,由O-TSa1中的263.36°,增加到OINTa1的287.86°,从而使碱基与糖环间的相对位置由反式变为了顺势.
途径四 在第四条反应途径中,水分子从糖环的上方进攻异头碳C1'原子并且由8-氧鸟嘌呤的O8原子摘取去氧糖糖环的Ha2',这条反应途径意味着异头碳C1'的结构保持.从图6中可以看出,在这种情况下,反应的第一步通过将去氧核糖糖环的Ha2'质子转移到碱基的O8原子产生一个类二氢呋喃中间体.第二步包括水分子加成到C-C双键上,而势能剖面图和各条反应通道的反应物、过渡态、中间体和产物的结构见图6.
水分子先与受损的去氧鸟苷作用形成8-氧去氧鸟苷-水复合物Rb,我们把Rb作为该反应通道的反应物.紧接着N-糖苷键发生扭转,使二面角O4'-C1'-N9-C4逐渐增大,在反应物中为257.81°,增大到第一个过渡态O-TSb1中的274.36°,从而使碱基与糖环间的相对位置由反式变为了顺势;同时N-糖苷键逐渐拉长,由反应物中的1.456到O-TSb1中的2.715,从图中可看出,OTSb1结构的N9-C1'键已经断裂,且键级为零.而Ha2'氢原子与OW间的距离以及HaW与O8原子的距离也逐渐缩短,分别由反应物中的2.626Å和3.808Å 缩短到 O-TSb1中的1.365Å 和1.272 Å,键角 OW-HaW-O8和 C2'-Ha2'-OW分别为168.91°和179.85°.
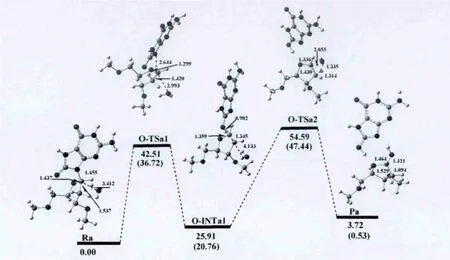
图5 以途径三进行的8-氧-2′-去氧鸟嘌呤核苷水解吉布斯自由能剖面图(kcal/mol)和优化结构及部分结构参数(键长:Å)Fig.5 Gibbs free energy cross-section(kcal/mol)of 8-oxo-2'-deoxyguanosine hydrolysis according to the third pathway and optimized structures along with their selected configuration parameters(bond lengths(inÅ)).
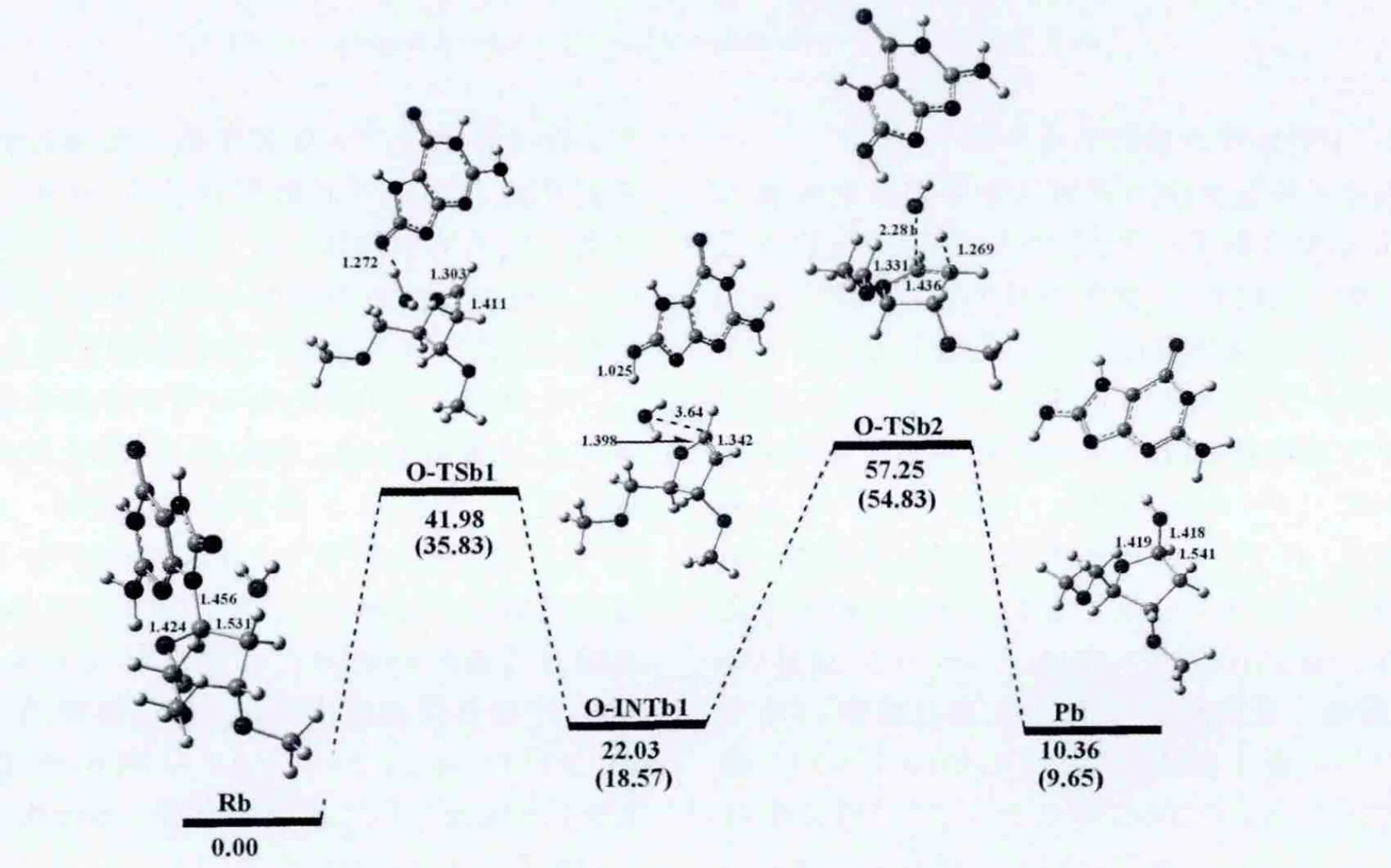
图6 以途径四进行的8-氧-7,8-二氢-2′-去氧鸟嘌呤核苷水解吉布斯自由能剖面图(kcal/mol)和优化结构及部分结构参数(键长:Å)Fig.6 Gibbs free energy cross-section(kcal/mol)of 8-oxo-2'-deoxyguanosine hydrolysis according to the fourth pathway and optimized structures along with their selected configuration parameters(bond lengths(inÅ)).
而从O-TSb1到第一个中间体O-INTb1的过程中,O8原子与HaW质子相结合,而OW与Ha2'质子相结合.二面角 O4'-C1'-N9-C4再逐渐减小,由 O-TSb1 中的 274.36°,减小 到 O-INTb1 的256.30°,从而使碱基与糖环间的相对位置再由顺式变回原来的反势;碱基环与新形成的水分子之间发生弱相互作用,OW与碱基上O8原子的HaW(由亲核试剂水分子转移到碱基上的质子)的距离为1.598 Å,键角 OW-HaW-O8 为 152.55°,且NBO分析表明,OW的孤对电子供给HaW-O8反键轨道的二阶稳定化能为37.79kcal/mol.
3.2 NBO分析
键级分析 键级的变化反映了化学键强度的变化,它与化学反应有着密切的联系,反映了化学反应的实质.对于该模型体系的所有驻点,我们在B3LYP/6-31++G(d,p)水平上计算所得的部分键级分别列于表1中.从表1中可以发现,对于途径一,反应物中的C1-C2键和O4'-C1键正常成键,其键级分别是0.9922和0.9012,到第一个过渡态时,由于N-糖苷键断裂及C2'-H的削弱,C1'原子与C2'原子间的相互作用和C1'原子与O4'原子间的相互作用增加,键级分别增加到1.1302和1.3898,已具有部分双键性质.最后在第一个中间体中,由于N-糖苷键断裂及C2'-H键的完全断裂,C1'原子与C2'原子间的相互作用进一步增加,C1'-C2'键的键级增加到1.8171,且成键轨道分析表明,C1'-C2'键确实包括一个σ键和一个π键,是典型的双键;由于C1'-C2'键双键的形成,C1'原子与C2'原子间的相互作用进一步增加,从而C1'原子与O4'原子间的相互作用减小,使其键级减小到1.0091,恢复到单键.而反应物、过渡态和中间体中C1'-C2'键的键长分别为1.537Å、1.452 Å和1.339Å,O4'-C1'键的键长分别为1.437Å、1.275Å和1.371Å,这与其键级的变化是一致的.其他反应途径的相应键长和键级的变化与途径一的相似.
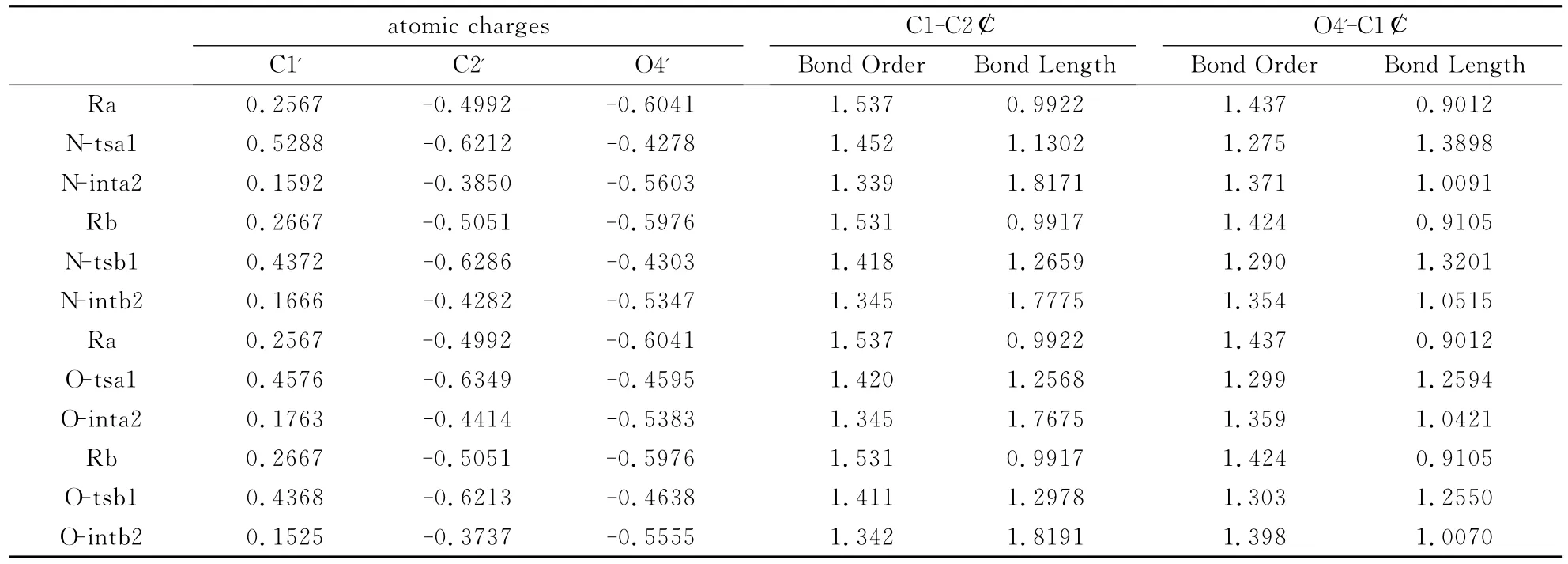
表1 8-氧-2′-去氧鸟嘌呤核苷水解过程中部分物质结构参数(原子净电荷:a.u.;键长:Å;键级)Table 1 Selected configuration parameters of 8-oxo-2'-deoxyguanosine hydrolysis.(atomic charge(in a.u.),bond length(inÅ)and bond order(in a.u.))
电荷分析 原子净电荷的变化反映了该原子在化学反应过程中所起的作用,甚至是直接参与成键、断键,它与化学反应有着密切的联系,反映了化学反应的实质.对于该模型体系的所有驻点,我们在B3LYP/6-31++G(d,p)水平上计算所得的部分原子净电荷分别列于表1中.从表1中可以发现,对于途径一,反应物中的C1'原子、C2'原子和 O4'原子的原子净电荷分别为0.2567、-0.4992和-0.6041.随着反应的进行,三个原子的原子净电荷也发生了改变.在第一个过渡态中,由于N-糖苷键断裂是以异裂过程进行,异头碳C1'原子带正电荷,有利于O4'原子的孤对电子离域到C1'原子的空轨道,从而使O4'原子的原子净电荷减小到-0.4278,而C1'原子的原子净电荷增加到0.5288.由二阶稳定化能分析发现,由O4'孤对电子C1'的p空轨道的稳定化能为5.71kcal/mol.又由于C2'-H的削弱,使C2'原子的原子净电荷增加到-0.62116.最后在第一个中间体中,由于C2'-H键的完全断裂,Ha2'以质子形式脱离糖环,使C2'原子的负电荷增加,从而C1'原子与C2'原子间的相互作用进一步增加,正负电荷部分抵消,结果是C1'原子与C2'原子间形成双键,且C1'原子与C2'原子的净电荷分别降低到0.1592和-0.3850.由于C1'=C2'双键的形成,C1'原子的净电荷降低,则O4'原子的孤对电子离域到C1'原子的空轨道的能力减低,结果O4'原子的原子净电荷又增加到-0.5603.用相同的方法可以分析其他反应途径的相应原子净电荷的变化,可得出与途径一相似的结论.
其实,这四个反应途径中,C1'原子上的正电荷和 O4'-C1'键 及 C1'-C2'键的缩短都可以用过渡态中去氧核糖糖环的氧碳正离子性质来解释.从图3—图6中可看出,这四条反应途径中,第一个过渡态结构的N9-C1'键已经断裂,键级为零.因此,在碱基和去氧核糖糖环之间存在重要的电荷分离.在反应途径一中,整个8-鸟嘌呤和去氧核糖糖环的电荷分别为-0.857和+0.850,而在反应途径二中,则分别为-0.752和+0.743,在反应途径三中,则分别为-0.700和+0.717,在反应途径四中,则分别为-0.733和+0.704.
3.3 能量分析
根据在B3LYP/6-31++G(d,p)水平上计算所得的相对能量的变化趋势所描绘的能级示意图如图3-图6所示.从图中可以发现,四条反应途径中,第一步即糖苷键断裂的相对能量最高,分别为49.34、47.31、42.50和41.99kcal/mol.第三条和第四条反应途径的活化能垒相近,只相差0.51kcal/mol;而第一条和第二条反应途径的活化能垒相近,相差2.03kcal/mol,且二者所需活化能较高.因此,从动力学上看,第三条和第四条反应途径比另外两条途径具有优势,反应主要通过途径三和途径四进行水解.
另一方面,这两种进攻方式的重要区别在于水分子在反应过程所起的作用不同.对N9原子摘取质子的反应而言,在途径一中,水分子没有直接参与到反应中,N9原子直接从去氧核糖糖环的C2'原子摘取 Ha2'质子,然而,在途径二中,C2'原子的Ha2'质子迁移到水分子,而后水分子的另一个质子再迁移到N9原子.后者是一种“溶剂辅助”效应,与反转方式相比较,降低活化能垒大约2 kcal/mol.这个过程降低能垒的程度不如其他过程降低活化能的程度大[16],因为这一步所需的能量主要作用于N-糖苷键的裂解.
由于真实的水解反应都是在溶剂中进行的,所以对于模型反应我们也考虑了溶剂化效应.我们选取水作为溶剂,在PCM/B3LYP/6-31++G(d,p)//B3LYP/6-31++G(d,p)水平上计算了各驻点的单点能,其相对能量变化如图3-图6所示.从图中可以看出,溶剂水对上述反应的溶剂化效应不明显,其相对自由能有所下降,但总的反应趋势没有发生改变,与气态中的反应趋势相同.所以,在水溶剂中,途径三和途径四仍然是优势途径.
4 小 结
本文用密度泛函DFT方法研究了N-糖苷键水解的反应机理.对模型体系采用了B3LYP方法在6-31++G(d,p)基组上对所有驻点进行了全优化.为了更好的理解水解反应机理,我们还研究了反应过程中键级和原子净电荷的变化、溶剂水对反应的溶剂化效应.研究结果表明,从水分子的进攻方向来看,8-oxodG与单个水分子作用的水解反应中,水分子有两个进攻方向,即水分子可以从去氧核糖糖环的上方和下方进攻异头碳C1'.结果,当水分子从去氧核糖糖环的下方进攻异头碳C1'时,异头碳C1'反应前后构型发生反转,而当水分子从去氧核糖糖环的上方进攻异头碳C1'时,异头碳C1'反应前后构型保持不变.另一方面,8-氧鸟嘌呤碱基的O8原子和N9原子都可以摘取去氧核糖糖环的 Ha2'.因此,8-oxodG与单个水分子作用的水解反应有四条不同的反应通道,且每条反应通道都包括两步,都形成类双氢呋喃中间体.计算结果表明,O8原子摘取去氧糖糖环Ha2'的反应的两条反应途径的第一步的活化能相近,途径三的活化能为42.50kcal/mol,比途径四的高0.51kcal/mol;而N9原子摘取去氧糖糖环Ha2'的反应的两条反应途径的第一步的活化能也相近,反转过程的活化能为49.34kcal/mol,比保留过程的活化能(47.31kcal/mol)高2.03kcal/mol.
由于N-糖苷键异裂的裂解能太高,N-糖苷键水解所需的活化能仍然相当高,因此,N-糖苷键的水解需要有催化剂的参与,而在人体内,存在各种酶参与N-糖苷键的水解.事实上,类二氢呋喃中间体的形成是一种保持生物体系内电中性的有利途径.
[1]Berti P J,McCann J A B.Toward a detailed under-standing of base excision repair enzymes:transition state and mechanistic snalyses of N-glycoside hydrolysis and N-glycoside transfer.[J]Chem.Rev.,2006,106(2):506.
[2]Mazumder D,Kahn K,Bruice T C.Computer simulations of trypanosomal nucleoside hydrolase:Determination of the protonation state of the bound transition-state analogue[J].J.Am.Chem.Soc.,2002,124(30):8825.
[3]Versees W,Barlow J,Steyaert J.Transition-state complex of the purine-specific nucleoside hydrolase of T.vivax:enzyme conformational changes and implications for catalysis[J].J.Mol.Biol.,2006,359(2):331.
[4]Roday S,Amukele T,Evans G B,etal.Inhibition of ricin A-chain with pyrrolidine mimics of the oxacarbenium ion transition state[J].Biochemistry.2004,43(17):4923.
[5]Osakabe T,Fujii Y,Hata M,etal.Targeting the undruggable proteome:the small molecules of my dreams[J].Chem-BioInf.J.,2004,4(1):73.
[6]Zaika E I,Perlow R A,Matz E,etal.Substrate discrimination by formamidopyrimidine-DNA glycosylase:a mutational analysis[J].J.Biol.Chem.,2004,279(6):4849.
[7]Bruner S D,Norman D P G,Verdine G L,etal.Structural basis for recognition and repair of the endogenous mutagen 8-oxoguanine in DNA[J].Nature,2000,403:859.
[8]Laudo M D,Whittleton S R,Wetmore S D M,et al.The effects of hydrogen bonding on the acidity of uracil[J].J.Phys.Chem.A,2003,107(46):10406.
[9]Petersson G A,Al-Laham M A.A complete basis set model chemistry(II):Open-shel1systems and the total energies of the first-row atoms[J].J.Chem.Phys.,1991,94(9):6081.
[10]Fukui K.A formulation of the reaction coordinate[J].J.Phys.Chem.,1970,74(23):4161.
[11]Reed A E,Curtiss L A,Weinhold F.Intermolecular interactions from a natural bond orbital,donoracceptor viewpoint[J].Chem.Rev.,1988,88(6):899.
[12]Barone V,Cossi M.Quantum calculation of molecular energies and energy gradients in solution by a conductor solvent model[J].J.Phys.Chem.A,1998,102(11):1995.
[13]Cossi M,Rega N,Scalmani G,Barone V.Energies,structures,and electronic properties of molecules in solution with the C-PCM solvation model[J].Comput.Chem.,2003,24(6):669.
[14]Frisch M J,Trucks G W,Schlegel H B,etal.Gaussian03D.01.Pittsburgh,PA:Gaussian,Inc.:2005.
[15]Zoltewicz J A,Clark D F,Sharpless T W,etal.Kinetics and mechanism of the acid-catalyzed hydrolysis of some purine nucleosides[J].J.Am.Chem.Soc.,1970,92(6):1741.
[16]Constantino E,Solans-Monfort X,Sodupe M,et al.Basic and acidic bifunctional catalysis:application to the tautomeric equilibrium of formamide[J].J.Chem.Phys.,2003,295(2):151.
