新生儿2q37缺失伴4q部分三体全基因组拷贝数变异(CNVs)分析
2014-05-07詹国栋黄国英周文浩
胡 萍 杨 琳 詹国栋 马 端 黄国英 周文浩
(1复旦大学附属儿科医院新生儿科-卫生部新生儿疾病重点实验室-上海市出生缺陷防治重点实验室 上海 201102;2复旦大学基础医学院-分子医学教育部重点实验室 上海 200032)
2号染色体末端缺失较为罕见,多数新发突变,极少数报道病例表现为亲代平衡易位缺失重复综合征的一部份[1]。2q37缺失可导致短指/趾-智力迟缓综合征(Brachydactyly-mental retardation syndrome,BDMR,OMIM 编号600430),也被称为Albright遗传性骨营养不良样综合征(Albright hereditary osteodystrophy-like syndrome,AHO-like syndrome),临床表现以特征性面部畸形、短缩指掌/趾跖骨(第3、4、5指趾受累明显)、运动及智力发育不同程度落后并伴有自闭症谱系为主要表现,是一种罕见染色体疾病[2-6]。1989年,Gorski等[7]首次报道了1例2q37缺失男孩,到目前为止国内外文献相关报道仅115例[8]。
4q部分三体综合征亦属罕见,1972年Surana和Conen[9]报道了第1例4q部分三体型的患者,随后Cervenka等[10]通过对另外几例患者临床表型的分析总结提出了4q部分三体综合征(Partial 4q trisomy syndrome)以来,至今全世界范围内病例数仅80余例。
与2q37缺失综合征不同的是,绝大多数4q部分三体型都是由于亲代平衡易位的不平衡分离染色体易位重排而来,少数为突变型,多数病例出生1年内死亡。由于几乎4号染色体长臂所有区段都参与了4q部分三体综合征的表现,其断裂点多变,常伴随其他单体型,加之分子遗传学精确基因定位较缺乏、现有报道患者年龄段散发,这些因素均导致明确该综合征基因型表现型的关系变得十分复杂和困难[11-12]。不伴其他染色体异常的情况通常被描述为“纯”4q部分三体综合征,其对于分析该综合征的基因型和表现型之间的关系,或是阐明某些特殊区段在表型产生中的作用,可能具有更重要的意义[13],这类型病例目前报道约30余例[13-18]。
本研究通过对1例新生儿多发畸形患儿进行全基因拷贝数变异分析,首次诊断了1例临床难以确诊的2q37缺失综合征合并4q部分三体综合征复杂染色体变异患儿。回顾国内外文献,尚未发现同时合并以上2种综合征基因型的相关报道,结合本例临床表型,与2种综合征分别进行对比研究,以期为完善相关基因型表型之联系丰富信息。
资料和方法
病例 复旦大学附属儿科医院新生儿病房收治的1例染色体核型分析提示为46,XX,add(2)(q37),2号染色体长臂异常的患儿(图1)。
伦理学 本研究经复旦大学附属儿科医院伦理委员会批准,患儿家属签署知情同意书后进入本项研究。患儿家长未同意进行家属的分子遗传学检测。
全基因组拷贝数变异检测 取外周静脉血经EDTA抗凝,使用QuickGene Mini80及DNA全血试剂盒(日本FUJI公司),从全血样本中提取DNA。使用NanoDropND 1000分光光度计检测DNA浓度及吸光度值,1%琼脂糖电泳检测DNA降解程度。取基因组DNA 3~5g进行芯片检测。采用全基因组2.7M芯片(美国Affymetrix公司),DNA样本送公司进行芯片检测。采用Chromosome Analysis Suite(ChAS)软件进行数据分析。

图1 患儿染色体核型分析结果Fig1 Chromosome karyotype analysis of the patient
罕见潜在临床意义CNVs片段的筛选 具有潜在临床意义的CNVs片段的筛选步骤:(1)ChAs软件分析的结果中,选择重复片段>150kb,缺失片段>50kb的CNVs数据行下一步分析;(2)对照原始图像除外假阳性结果;(3)除外在国际基因组拷贝数变异多态性数据库(Database of Genomic Variation,DGV)报道的正常人群的CNVs(片段至少有80%以上重合,且重复或缺失类型相同);(4)除外片段区域内不包含基因的CNVs;(5)选取常染色体上>2.5Mb的CNVs。
结 果
患儿临床资料 女,1天,G4P2,其母30岁,孕37+5周因胎动少、瘢痕子宫行剖宫产娩出;出生体重2700g,羊水清,无胎膜早破,Apgar评分:1min,8分;5min,9分。因“生后发现外耳畸形”入住复旦大学附属儿科医院。查体:头围34cm,身长48cm,神志清,哭声低;小耳畸形,外耳廓卷曲,小下颌,颈蹼,双手通贯掌,双足内翻;四肢肌张力可,原始反射弱。超声心动图示室间隔(肌部)缺损,动脉导管未闭,卵圆孔未闭,肺动脉高压,二尖瓣(降落伞部)狭窄。胸腹平片示胸廓略显狭长,心影饱满,脊柱后凸畸形。腹部B超示胆囊内液混,左肾盂轻度增宽。头颅MRI示双侧侧脑室饱满,右额叶异常信号,白质损伤可能。余未见异常。其母第1胎在胎龄5个月时因发现颈部肿物高度怀疑畸形行人工流产,第2胎死产,另有一子,3岁、体健。父母体健,否认家族中糖尿病、先天性心脏病史,否认近亲结婚。母亲否认孕期感染史、致畸剂接触史和传染病接触史。
全基因组拷贝数变异基因芯片结果 表1为本例CNVs起始位点及大小。图2显示本例为2号染色体长臂的缺失伴4号染色体长臂的重复,缺失与重复的片段均为靠近长臂末端的较大片段。

图2 患儿2号染色体缺失并4号染色体重复基因芯片检查结果Fig2 Microarry results of the proband with deletion in chromosome 2and duplication in chromosome 4
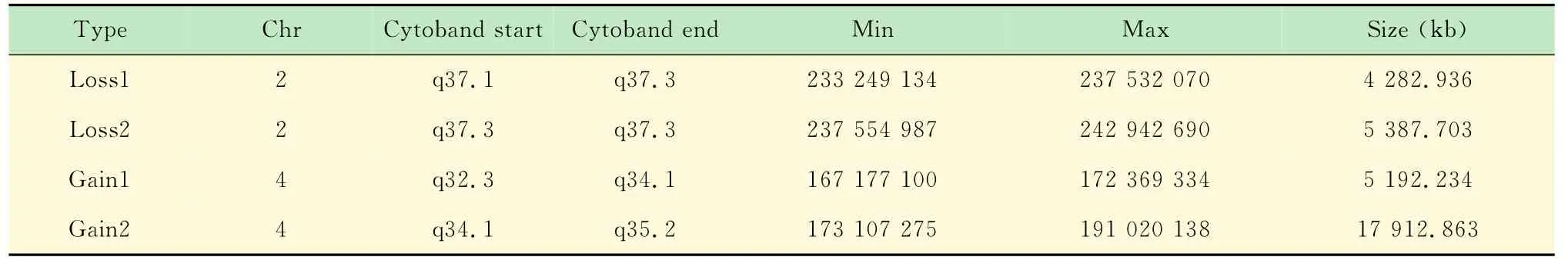
表1 本例患儿筛选出的CNVs基本特征Tab1 Characteristics of rare CNVs with potential clinical significance of the case
讨 论
基因芯片结果显示本例患儿为2q37缺失合并4q32-35重复的复杂型染色体异常,我们将患儿表型与相应综合征报道表型对比分析如下。
2q37缺失典型面部特征在本例中的表现 典型的面部畸形特征包括圆脸、前额突出、眉毛弓形高挑、睑裂下斜、V字形鼻、鼻梁低平、上腭高、毛发细疏、耳部发育不良、人中短而平、上唇薄等[19]。本例患儿面部特征仅表现为小耳畸形、耳廓卷曲及小下颌,考虑原因:(1)患儿处于新生儿期,于其他报道年长患者相比,面部特征有待发育;(2)患儿合并其他染色体异常;(3)2q37.3亚端粒区缺失在正常人群存在多态性[20-21],在患者中也有异质性;(4)典型面部畸形特征虽然占报道病例的30%~50%[3,22]左右,但仍有部分患儿不表现出此典型的面部特征。
2q37缺失综合征的分型在本例中的体现 另外Shrimpton等[23]通过对比文献总结发现,2q37缺失综合征根据临床表现可以划分为两个类别:其一是多数病例报道的中到重度精神运动发育迟缓伴多形非特异性先天畸形。除了以上讨论的面部畸形之外,还包括20%[19]左右病例表现为心血管系统畸形,特征性的骨骼发育畸形,比例不等的病例表现为泌尿生殖系统、中枢神经系统和呼吸消化系统畸形;精神运动发育迟缓则表现为肌张力低下、智力障碍、自闭症谱系行为异常等问题,占到了报道病例的30%以上。其二为少数病例表现为轻度智力障碍伴明显的AHO-like综合征,主要临床特征为发育迟缓、身材短小、肥胖倾向、短缩指掌/趾跖骨(并指、指弯曲等亦属常见)等,类似于Albright遗传性骨营养不良综合征(假-假性甲状旁腺功能减退症)。结合本例患儿小耳小下颌面部畸形、心血管系统异常、脊柱后凸骨骼畸形以及脑部、肾脏、胆囊等影像学异常情况,我们可以初步判断这是一个属于多数情况的2q37缺失综合征。
2q37缺失综合征致病关键基因与本例表型的关系 由于2q37缺失综合征的表型变异性大,虽然报道的病例数已超过百余例,但多数病例源于2q37末端缺失,不利于关键基因的定位;因此关于2q37缺失综合征的基因型表型之间的关联尚未完全明确,既往研究将关键区域定位在 2q37.1-qter[4,24]。近年来才发现致病基因HDAC4(histone deacetylase 4),该基因为II型组蛋白脱乙酰酶,定位在2q37.3,主要作用为抑制转录因子RUNX2和MEF2C,两者均为骨骼发育必需的转录因子蛋白。另外该基因还通过调节一系列与肌肉、心脏及神经发育相关的蛋白因子发挥作用[25-27],分别影响BDMR各系统的主要表型特征。
Vega等[28]2004年的小鼠动物实验研究表明,基因敲除HDAC4的小鼠在早期软骨发育中表现出严重的骨骼畸形,说明单个HDAC4的基因突变或缺失即可引起BDMR特征性指跖骨畸形的产生。Morris等[29]报道了BDMR家系1例,亲代症状轻、子代症状重,基因分析发现HDAC4的表达在两代人中有显著差别,分别是67%和23%;阐明该基因呈剂量依赖性调节BDMR综合征的表型严重程度。结合本例,患儿存在HDAC4的基因缺失,但是没有明显的指跖骨特征性短缩畸形,考虑原因:(1)该表型虽为BDMR特征性表型,但只存在于50%~60%的病例中;(2)另外 Villavicencio-Lorini等[30]最近对同一家系内3代2q37.3缺失患者的研究发现,HDAC4的单倍不足导致了该家系患者不同程度的精神运动行为等异常合并特殊面部畸形的发生,但是这3代患者均无特征性指跖骨短缩畸形,作者推测HDAC4基因单倍不足有可能存在补偿机制;(3)患儿2q37为非单纯性缺失,且同时存在2q37.1和2q37.3两个基因断裂点,基于2q37区域为一基因丰富区段,此断裂极有可能干扰了邻近基因的协同或拮抗作用;(4)进一步说明本例患儿可划分为不属于AHO-like表现型一类的非典型2q37缺失综合征。
本例与具有类似区段重复的4q部分三体综合征病例比较 文献资料表明,肾脏畸形伴或不伴拇指异常是4q部分三体典型的临床表现,其与4q三体区段的对应关系可能涉及4q22~q23[31]、4q25~q28[12]、4q33~q34[16]。结合本例患者存在左侧肾盂轻度增宽的影像学表现及4q32.3~q34.1区段重复遗传学表现,同时本例患者亦为女性,进一步支持Otsuka等[16]肾脏发育不良女性多发的观点。
Elghezal等[32]则表明4q35区段很有可能参与了小头畸形、严重生长发育及智力迟缓的发生;然而近年Kim等[33]则报道了1例35岁男性4q32~q35同臂插入重复,仅表现为不孕症状。2012年Martin等[34]则报道了一家系内4q32.1~q35.1区段倒置重复的病例,并通过涉及相关区段的“纯”4q部分三体综合征的病例对比发现4q33~q34是导致精神运动发育损害的关键区段。虽然本例包含该关键区段的重复,但由于患儿出生后3天即死亡,无法评估其精神运动发育情况。
从患儿双耳轮折叠、双手通贯掌、足内翻畸形、肾脏发育不良及心脏多发畸形等情况来看,与4q部分三体综合征对应的面部特征及肢端-心脏-肾脏异常症候群非常符合Carrascosa等[35]报道的1例4q31~q35重复的病例,并支持其结论即该区段也参与了4q部分三体综合征。另外类似的病例还有2005年 Otsuka等[16]报道的1对4q31.22~q35.2重复的兄妹,虽然2个先证者都缺乏文献[36]证实的与泌尿生殖系畸形相关的4q22~q23区段三体型,但均有明显肾脏发育不良。由于4q部分三体综合征的特征性拇指畸形相关区段被定位在4q27~q28[12],所以本例患儿虽有4q部分三体综合征多数表现,但是无此典型畸形特征。
2007年Senses等[15]报道了1例4号5号染色体易位导致4q31.3~qter重复的新生儿,与本例相似的表型有低耳位伴发育不良、小下颌、双手通贯掌及心脏畸形,作者总结了17例“纯”4q部分三体综合征表型,得出结论即易位的4q三体型临床表型与“纯”4q部分三体综合征趋于一致,由此支持我们的推断,即本例患儿表型极有可能是亲代平衡易位的不平衡分离而导致的基因组失衡。
2010年Egritas等[14]报道了1例4q25-qter区段重复的新生儿胆汁淤积症,患儿存在典型的拇指畸形、耳部发育不良、马蹄足内翻、短颈等4q部分三体型的常见表现,是目前唯一1例4q部分三体型当中报道有胆汁淤积症的病例。相比之下,本例患儿存在腹部B超示胆囊内液混的影像学表现和其他4q部分三体的常见表现,提示本例新生儿也有进展为胆汁淤积的可能,同时为4q部分三体基因型与新生儿胆汁淤积的表型关系丰富了区段信息。
4号染色体关键致表型基因在本例患儿中的体现 由于4q2-q3不同区段共同参与了4q部分三体综合征,且该区段所包含基因丰富,目前多数研究都着眼于定位关键致病区段,相关致病基因与表型联系的精确定位研究尚未形成。目前仅有关于HDAN2基因的一项动物实验,Tamura等[37]表明该基因的剂量负荷是导致4q部分三体型当中心脏及肢端畸形的一个主要原因。结合本例患儿4q32.3~q34.1区段重复导致HDAN2基因过表达,产生明显的心脏畸形及双手通贯掌双足内翻畸形等肢端异常表型,支持这一三体区基因剂量失衡致病机制的解释。
小结 由于本例患儿生后第3天即死亡,缺少对其精神运动发育及智力发育等生长情况的随访评估,仅从患儿多发畸形的初期表现及相关文献的对比研究,我们推测此基因型的单体型和三体型对于患儿的临床表型都发挥了致病作用。我们将其与2种综合征的最具共性的临床表型进行对比(表2),同时也将部分已报道涉及相应区段的“纯”4q部分三体综合征进行了临床资料的横向比较(表3),发现4q重复对患儿表型的影响更大。考虑4q异常染色体片段更大,对本例表型的贡献程度可能更多,且患儿早期死亡也更符合4q部分三体综合征多数病例的结局;同时因为无法取得患儿双亲及其家系内同胞基因型,限制了我们对其染色体变异机制的进一步研究。本文首次初步建立了2q37缺失综合征合并4q部分三体综合征患儿这一十分罕见的基因型表现型关联信息,但是更精确的基因型表现型相关关系尚需积累相应的病例资料及相关基因功能研究来确证。

表2 本例患儿表型与两种综合征典型表现的比较Tab2 Comparation of the common clinical findings of the child and the two syndromes

表3 涉及本例患儿重复区段的“纯”4q部分三体综合征临床表型比较Tab3 Clinical findings in patients with pure duplication invovling 4q32.3-q35.2

(续表3)
[1]Schinzel A. Catalogue of unbalanced chromosome aberrations in man[M].2nd ed.New York:Walter de Gruyter Inc,2001.
[2]Galasso C,Lo-Castro A,Lalli C,et al.Deletion 2q37:an identifiable clinical syndrome with mental retardation and autism[J].J Child Neurol,2008,23(7):802-806.
[3]Casas KA,Mononen TK,Mikail CN,et al.Chromosome 2q terminal deletion:report of 6new patients and review of phenotype‐breakpoint correlations in 66individuals[J].Am J Med Genetics Part A,2004,130(4):331-339.
[4]Aldred MA,Sanford ROC,Thomas NS,et al.Molecular analysis of 20patients with 2q37.3monosomy:definition of minimum deletion intervals for key phenotypes[J].J Med Ggenetics,2004,41(6):433-439.
[5]Wilson LC,Leverton K,Luttikhuis MEMO,et al.Brachydactyly and mental retardation:an Albright hereditary osteodystrophy-like syndrome localized to 2q37[J].Am J Hum Genet,1995,56(2):400-407.
[6]Conrad B,Dewald G,Christensen E,et al.Clinical phenotype associated with terminal 2q37deletion[J].Clin Genetics,1995,48(3):134-139.
[7]Gorski JL,Cox BA,Kyine M,et al.Terminal deletion of the long arm of chromosome 2in a mildly dysmorphic hypotonic infant with karyotype 46,XY,del(2)(q37)[J].Am J Med Genetics,1989,32(3):350-352.
[8]Leroy C,Landais E,Briault S,et al.The 2q37-deletion syndrome:an update of the clinical spectrum including overweight,brachydactyly and behavioural features in 14 new patients[J].Eur J Hum Genetics,2013,21(6):602-612.
[9]Surana RB,Conen PE.Partial trisomy 4resulting from a 4-18reciprocal translocation[J].Ann Genet,1972,15(3):191-194.
[10]Cervenka J,Djavadi GR,Gorlin RJ.Partial trisomy 4q syndrome:case report and review[J].Hum Genetics,1976,34(1):1-7.
[11]Cernakova I,Kvasnicova M,Lovasova Z,et al.A duplication dup(4)(q28q35.2)de novo in a newborn[J].Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub,2006,150(1):113-116.
[12]Battaglia A,Chen Z,Brothman AR,et al.Karyotype/phenotype correlations in duplication 4q:Evidence for a critical region within 4q27-28for preaxial defects[J].Am J Med Genetics Part A,2005,134(3):334-337.
[13]Rinaldi R,Bernardo C,Assumma M,et al.Cytogenetic and molecular characterization of a de novo 4q24qter duplication and correlation to the associated phenotype[J].Am J Med Genetics Part A,2003,118(2):122-126.
[14]Egritas O,Cavdarli B,Dalqic B,et al.Duplication 4q associated with chronic cholestatic changes in liver biopsy[J].Eur J Med Genetics,2010,53(6):411-414.
[15]Senses D,Silan F,Uzun H,et al.Partial trisomy 4(q31qter)due to maternal 4,5balanced translocation in a neonate[J].Genetic Counseling,2007,18(2):163-170.
[16]Otsuka T,Fujinaka H,Imamura M,et al.Duplication of chromosome 4q:renal pathology of two siblings[J].Am J Med Genetics Part A,2005,134(3):330-333.
[17]Lin S,Kirk EP,McKenzie F,et al.De novo interstitial duplication 4(q28.1q35)associated with choanal atresia[J].J Paediatr Child Health,2004,40(7):401-403.
[18]Celle L,Lee L,Rintoul N,et al.Duplication of chromosome region 4q28.3-qter in monozygotic twins with discordant phenotypes[J].Am J Med Genetics,2000,94(2):125-140.
[19]Falk RE,Casas KA.Chromosome 2q37deletion:clinical and molecular aspects[J].Am J Med Genetics Part C:Semin Med Genetics,2007,145C(4):357-371.
[20]Macina RA,Negorev DG,Spais C,et al.Sequence organization of the human chromosome 2q telomere[J].Hum Mol Genetics,1994,3(10):1847-1853.
[21]Fan YS,Zhang Y,Speevak M,et al.Detection of submicroscopic aberrations in patients with unexplained mental retardation by fluorescence in situ hybridization using multiple subtelomeric probes[J].Genetics Med,2001,3(6):416-421.
[22]Reddy KS,Flannery D,Farrer RJ.Microdeletion of chromosome sub-band 2q37.3in two patients with abnormal situs viscerum[J].Am J Med Gnetics,1999,84(5):460-468.
[23]Shrimpton AE,Braddock BR,Thomson LL,et al.Molecular delineation of deletions on 2q37.3in three cases with an Albright hereditary osteodystrophy-like phenotype[J].Clin Genetics,2004,66(6):537-544.
[24]Chaabouni M,Le Merrer M,Raoul O,et al.Molecular cytogenetic analysis of five 2q37deletions:refining the brachydactyly candidate region[J].Eur J Med Genetics,2006,49(3):255-263.
[25]Darcy MJ,Calvin K,Cavnar K,et al.Regional and subcellular distribution of HDAC4in mouse brain[J].J Comp Neurol,2010,518(5):722-740.
[26]Chen B,Cepko CL.HDAC4regulates neuronal survival in normal and diseased retinas[J].Science,2009,323(5911):256-259.
[27]Zhang CL,McKinsey TA,Chang S,et al.Class II histone deacetylases act as signal-responsive repressors of cardiac hypertrophy[J].Cell,2002.110(4):479-488.
[28]Vega RB,Mstsuda K,Oh J,et al.Histone deacetylase 4 controls chondrocyte hypertrophy during skeletogenesis[J].Cell,2004,119(4):555-566.
[29]Morris B,Etoubleau C,Bourthoumieu S,et al.Dose dependent expression of HDAC4causes variable expressivity in a novel inherited case of brachydactyly mental retardation syndrome[J].Am J Med Genetics Part A,2012,158A(8):2015-2020.
[30]Villavicencio-Lorini P,Klopocki E,Trimborn M,et al.Phenotypic variant of Brachydactyly-mental retardation syndrome in a family with an inherited interstitial 2q37.3 microdeletion including HDAC4[J].Eur J Hum Genetics,2013,21(7):743-748.
[31]Lundin C,Zech L,Sjörs K,et al.Trisomy 4q syndrome:presentation of a new case and review of the literature[J].Ann Genet,2002,45(2):53-57.
[32]Elghezal H,Sendi HS,Monastiri K,et al.Large duplication 4q25-q34with mild clinical effect[J].Ann Genet,2004,47(4):419-422.
[33]Kim JW,Park JY,Oh AR,et al.Duplication of intrachromosomal insertion segments 4q32→q35 confirmed by comparative genomic hybridization and fluorescent in situ hybridization[J].Clin Exp Reprod Med,2011,38(4):238-241.
[34]Martin J,Saleki R,Christensen T,et al.Familial 25.3Mb inverted duplication of bands q32.1to q35.1on chromosome 4with psychomotor impairments[J].Am J Med Genetics Part A,2012,158(10):2624-2628.
[35]Carrascosa Romero MC, García Mialdea O, Vidal Company A,et al.Duplicación parcial del cromosoma 4q(q31,q35):síndrome aurículo-acro-renal[J].An Pediatr,2008,68(4):361-364.
[36]Zollino M,Zampino G,Torrioli G,et al.Further contribution to the description of phenotypes associated with partial 4q duplication[J].Am J Med Genetics,1995,57(1):69-73.
[37]Tamura M,Hosoya M,Fujita M,et al.Overdosage of Hand2causes limb and heart defects in the human chromosomal disorder partial trisomy distal 4q[J].Hum Mol Genetics,2013,22(12):2471-2481.
[38]Petriczko E,Biczysko-Mokosa A,Bogdanowicz J,et al.Familial distal monosomy 3p26.3-pter with trisomy 4q32.2-qter,presenting with progressive ataxia,intellectual disability,and dysmorphic features[J].Am J Med Genetics Part A,2012,158(6):1442-1446.
[39]Vogel W,Siebers JW,Gunkel B,et al.Uneinheitlicher Phänotyp bei Partialtrisomie 4q[J].Hum Genet,1975,28(2):103-112.
[40]Dutrillaux B,Laurent C,Forabosco A,et al.La trisomie 4q partielle.A propos de trois observations[J].Ann Genet(Paris),1975,18(1):21-27.
[41]Taylor KM,Francke U,Brown MG,et al.Inverted tandem(“mirror”)duplications in human chromosomes:inv dup 8p,4q,22q[J].Am J Med Genetics,1977,1(1):3-19.
[42]闭宏娟.新生儿4号染色体长臂部分三体综合征1例[J].临床荟萃,2012,27(015):1364-1364.
[43]Goodman BK,Capone GT,Hennessey J,et al.Familial tandem duplication of bands q31.1to q32.3on chromosome 4with mild phenotypic effect[J].Am J Med Genetics,1997,73(2):119-124.
