质谱非标记定量分析小麦抗条锈病近等基因系蛋白质变化
2014-03-17黄宇冯晶饶力群徐世昌潘映红
黄宇冯晶饶力群徐世昌潘映红
(1.湖南农业大学生物科学技术学院,长沙 410128;2.中国农业科学院作物科学研究所,北京 100081;3.国家农作物基因资源与基因改良重大科学工程,北京 100081;4.中国农业科学院植物保护研究所,北京 100193)
质谱非标记定量分析小麦抗条锈病近等基因系蛋白质变化
黄宇1,2,3冯晶4饶力群1徐世昌4潘映红2,3
(1.湖南农业大学生物科学技术学院,长沙 410128;2.中国农业科学院作物科学研究所,北京 100081;3.国家农作物基因资源与基因改良重大科学工程,北京 100081;4.中国农业科学院植物保护研究所,北京 100193)
利用基于液相色谱串联Orbitrap质谱(Q Exactive)的非标定量(Label-free)技术,对小麦抗病单基因近等基因系Taichung29*6/Yr10和背景品种Taichung29叶片的蛋白质表达进行了比较分析。经MASCOT软件搜库,在两个样品中共同鉴定到2 257个蛋白,其中有准确定量信息的蛋白共1 549个,含量变化大于2倍的差异蛋白有102个。对102个差异表达蛋白进行了Gene Ontology(GO)功能注释和分析,发现他们多数定位于细胞基质和核糖体等细胞器,具备结合、催化等功能,主要参与代谢、细胞过程、应激等生物学进程,其中超氧化物歧化酶、甲硫氨酰氨肽酶、溶酶体-β-葡萄糖苷酶和铁蛋白等可能与小麦抗病单基因近等基因系Taichung 29*6/Yr10的抗病性有一定相关性。
小麦 条锈病 抗病性 蛋白质组学 非标记定量 Q Exactive质谱
小麦条锈病是由小麦条锈病菌(Puccinia striiformisf. sp.tritici)引起的小麦叶部病害,严重时也危害穗部。小麦条锈病在许多国家和地区均有发生,我国是小麦条锈病发生最广泛的国家之一[1,2]。防治小麦条锈病最有效、最经济、最安全的方法是利用抗病品种。虽然国际上已经命名了49个位点的55个小麦抗条锈病基因(Yr1-Yr52),但其中大多数抗病基因具有生理专化性,常常因为条锈菌小种的快速变异而失去抗性,目前,只有少数的抗病基因仍保持抗性,如Yr5、Yr10和Yr15等[2-5]。随着大
量物种基因组测序的完成,人们清楚的意识到单纯从基因组和转录组的信息并不能完全揭示生命活动的规律,作为功能基因组学研究内容之一的蛋白质组学已经成为生命科学研究领域的热点和前沿[6]。目前,已知的小麦抗条锈病基因都是通过遗传推导确定的,仅有极少数基因如Yr36的序列已经明确。因此,从蛋白质组学的角度去揭示病原物致病机理和抗病品种的抗病机制,对小麦抗条锈性的合理利用和小麦条锈病的可持续控制极具研究价值。
双向电泳是目前蛋白质组学研究的传统方法[7-9],但该技术本身存在分离蛋白范围有限、歧视效应、与质谱联用效果差等诸多缺陷[10]。近年来,随着高精度生物质谱技术和基于生物信息学的海量数据处理技术的进步,规模化的精确测量细胞内蛋白质组的表达变化已成现实。定量蛋白质组研究方法也从传统的基于二维凝胶电泳结合质谱鉴定的策略向着更高准确度、更大范围和更高通量的方向发展。蛋白质组学定量方法根据是否对蛋白质/多肽进行标记可以分为标记和非标记两类。标记定量方法是基于在肽段中引入稳定同位素标记(如2H、13C、15N、18O),使得在同一次质谱扫描中标记“轻”和“重”同位素的多肽具有相同的色谱行为和离子化效率,此时成对出现的质谱峰信号的相对强度可以精确地反映样品中蛋白质的比例。根据同位素掺入的方式,标记方法可以分为代谢标记、酶促标记和化学标记等,在这些标记方法中,化学标记的种类最为繁多,应用也最为广泛。代表性的化学标记方法包括标记多肽(蛋白质)N端氨基的iTRAQ[11]、标记C端羧基的酯化标记、针对半胱氨酸巯基的ICAT[12]以及与质谱多反应监测(MRMMS)技术联用的mTRAQ标记技术[13]等。以上这些标记定量方法具有良好的定量可靠性,不仅可以用于不同样品间的相对定量分析,还可以用于蛋白质的绝对定量分析,但成本普遍偏高。相对于传统的标记定量法,非标定量法(Label-free)不需要在样本分析前对蛋白/多肽进行标记,避免了样品处理过程中可能造成的损失,在检测肽段的数量、蛋白质的覆盖率和分析通量方面具有较大优势,且不受样品来源和数量限制。非标定量法通过比较质谱谱图计数(spectrum counting)或质谱峰强度(peak area intensity)分析不同来源样品蛋白的含量变化,是目前比较蛋白质组学上运用较为普遍的一种手段。Boeri等[14]采用Label-free方法和MALDI-Q-TOF 技术在定量分析表皮生长因子的磷酸化蛋白动力学、蛋白质覆盖率、序列鉴定、识别磷酸化位点等方面取得了新进展。Wang等[15]采用非标定量方法分别结合LCQ、LTQ-FT质谱对多种样品的重现性作了测试,并详细研究分析了蛋白质的磷酸化过程。最近,Zhang等[16]借助LTQ质谱和PTMap软件,鉴定出酵母菌组蛋白上新的丙酰化和丁酰化修饰,并发现了14种潜在的、未知的蛋白修饰。
本研究以非标定量法结合液相色谱串联质谱技术对小麦感病品种Taichung29(T29)以及抗病近等基因系Taichung 29*6/Yr10(TYr10)进行差异蛋白分析,并通过生物信息学的方法分析差异蛋白所属的细胞成分(Cellular component)、分子功能(Molecular function)和生物学途径(Biological process),旨在为小麦条锈病的可持续控制研究提供线索。
1 材料与方法
1.1 材料
1.1.1 材料来源 小麦感病品种T29以及抗病品种近等基因系TYr10小麦均由中国农业科学院植物保护研究所条锈病组繁殖培养。
1.1.2 材料的培养 小麦种子经浸润,催芽后种于口径50 cm×100 cm塑料盆中,置于自控温室中(温度昼15-20℃/夜11-14℃,光照强度6 000 Lx,光照时间12 h),直到幼苗于温室内培养至1叶期。
1.1.3 主要仪器和试剂 HPLC液相系统Easy-nLC 1000(美国Thermo Scientific公司);Q Exactive MS质谱仪(美国Thermo Scientific公司);Mini垂直蛋白电泳仪(美国Bio-Rad公司);Milli-Q超纯水系统(美国Millipore公司);冷冻高速离心机(美国科峻仪器公司);Lambda25型紫外可见分光光度计(美国 Perkin-Elmer 公司);溴酚蓝bromophenol blue、碘乙酰胺IAA、二硫基苏糖醇DTT、聚丙烯酰胺Acr、过硫酸钠APS、甘氨酸Glycine、四甲基乙二胺TEMED、十二烷基硫酸钠SDS、三羟甲基氨基甲烷Tris(美国GE Healthcare公司);测序级胰蛋白酶TPCK-Trypsin(美国Promega公司);乙腈ACN、三
氟乙酸TFA(美国Fisher Scientific公司)。
1.2 方法
1.2.1 小麦叶片总蛋白的提取 根据刘伟霞等[17]的方法稍加修改,称取新鲜1叶期小麦叶片2 g,液氮研磨20 min,转入50 mL离心管中,加入50 mL预冷丙酮(2% DTT,10% TCA)-20℃过夜沉淀(至少12 h)。离心40 000×g,1 h,弃去上清。加入50 mL冰丙酮(2% DTT)放入-20℃,静置1 h(中间震荡3次)40 000×g,1 h离心,重复3次,将沉淀至于通风厨吹干。沉淀即为蛋白粗样品,若要长期储存则放于-80℃冰箱保存,或将干粉溶于UA缓冲液中(尿素8 mol/L,Tris-HCl 100 mmol/L,pH8.5)用于电泳以及酶解。
1.2.2 蛋白定量 参照Bradford法进行蛋白定量[18],采用BSA做标准曲线。
1.2.3 SDS-PAGE 1D SDS-PAGE参考仪器说明并在Mini-protean Ⅲ cell垂直板电泳仪上进行。
1.2.4 FASP酶解 取蛋白总量为1 mg的蛋白裂解样品于10 k超滤管中,并加入DTT(20 mmol/L)还原3 h,加入200 μL UA缓冲液,离心14 000×g,15 min离心,重复两次。加入100 μL IAA,600 r/min混匀1 min,暗处孵育20 min,14 000×g,10 min离心。加入200 μL UA,再离心15 min,重复两次。加入200 μL ABC(碳酸氢铵0.05 mol/L),离心14 000×g,10 min离心,重复两次。转入新的收集管,加入一定量ABC,加入Trypsin(1∶50);混匀,37℃摇床16-20 h离心14 000×g,10 min离心,加入40 μL 50 mmol/L ABC,离心14 000×g 10 min离心,冻干,-20℃保存。
1.2.5 色谱条件 高效液相色谱仪:Thermo Scientific Easy-nLC 1000;色谱柱:Easy-spray column(C18,2 μm,100Å,75 μm×15 cm);流动相:A:0.1% Formic acid,2% Acetonitrile in water;B:0.1% Formic acid in Acetonitrile;梯度:3%-8% B in 10 min,8%-20% B in 78 min,20%-30% B in 15 min,30%-90% B in 10 min,90% B for 7 minutes;流速:250 nL/min。
1.2.6 质谱分析条件 质谱仪:Thermo Scientific Q Exactive;喷雾电压:2.3 kV;毛细管温度:250℃;S-lens:55%;碰撞能量:27% HCD;分辨率设置:一级70 000@m/z 200,二级17 500@m/z 200;母离子扫描范围:m/z 300-1800;子离子扫描范围:start from m/z 100;Data-dependent MS/MS:Top 15。
1.2.7 Q Exactive质谱数据分析 原始文件(raw file)使用Maxquant软件进行Label-free定性和定量分析,搜索相应的小麦蛋白数据库。具体搜库参数如下:前体离子质量偏差:10 ppm;碎片离子质量偏差:25 mmu;固定修饰:半胱氨酸(Cysteine)烷基化(+57.021 Da);动态修饰:甲硫氨酸(Methionine)氧化(+15.995 Da);天冬酰胺和谷氨酰胺(Asparagine & Glutamine)脱氨基化(+0.984 Da);酶:trypsin;漏切位点:2。数据库来自UNIPROT(http://www. uniprot.org/)。
1.2.8 差异表达蛋白的GO(Gene Ontology)分析根据蛋白数据库中的各蛋白GO注释,并结合其来源,人工选择鉴定所得到的蛋白最可能的细胞成分、分子功能和生物学途径,并对其进行基因富集度计算和图示化。鉴定蛋白的GO信息来自UNIPROT网站(http://www.uniprot.org/)。
2 结果
2.1 小麦叶片总蛋白的提取分析与SDS-PAGE电泳
小麦叶片通过TCA/丙酮沉淀,用UA裂解液溶解,通过Bradford法进行定量,测出TYr10中蛋白浓度为7.80 mg/mL,T29提取蛋白浓度为8.01 mg/mL,计算得出总蛋白的提取率分别为0.58%和0.60%。通过SDS-PAGE(图1)对提取结果进行检测发现,两组样品之间平行度较好,相应的蛋白条带清晰,初步表明蛋白提取效果理想,样品纯度适于进行下一步工作。

图1 小麦叶片总蛋白SDS-PAGE电泳图
2.2 液相色谱串联质谱鉴定
将蛋白粗样品复溶、酶切之后,经过Easy-nLC 1000液相系统进行色谱分离,并通过Q Exactive进行质谱分析(3次技术重复),得到的数据进行Mascot搜索,共鉴定到2 257个蛋白。对已鉴定蛋白通过严格标准进行筛选(peptide FDR & protein FDR≤1%),且设定每个蛋白需有两条以上肽段提供定量比值,最终确定具有可靠定量信息的蛋白有1 549个(数据略)。
2.3 差异表达蛋白筛选结果及其GO分析
在上述具有定量信息的1 549个蛋白中,共筛选出蛋白表达水平差异大于2倍,且3次技术重复RSD<50%的蛋白102个,其中低表达31个,高表达71个(表1)。这些蛋白的相对分子量(MW)分
布为4.42 kD(D2K937)-164.95 kD(M7ZSA1),等电点(pI)分布为4.71(M7Z596)-12.01(Q95H53),相对分子量和理论等电点分布广泛。
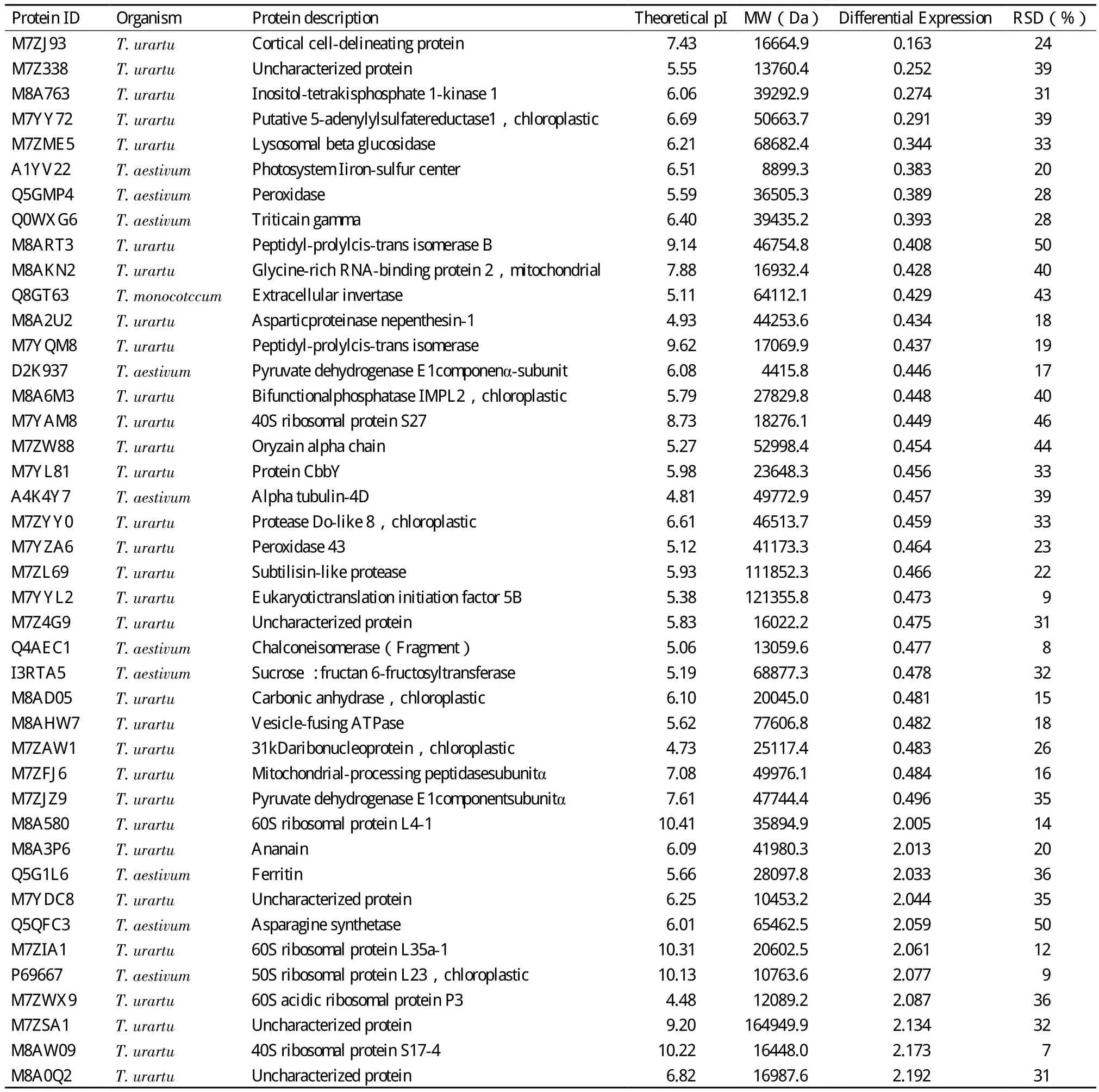
表1 质谱数据分析差异蛋白结果(TYr10 vs T29)
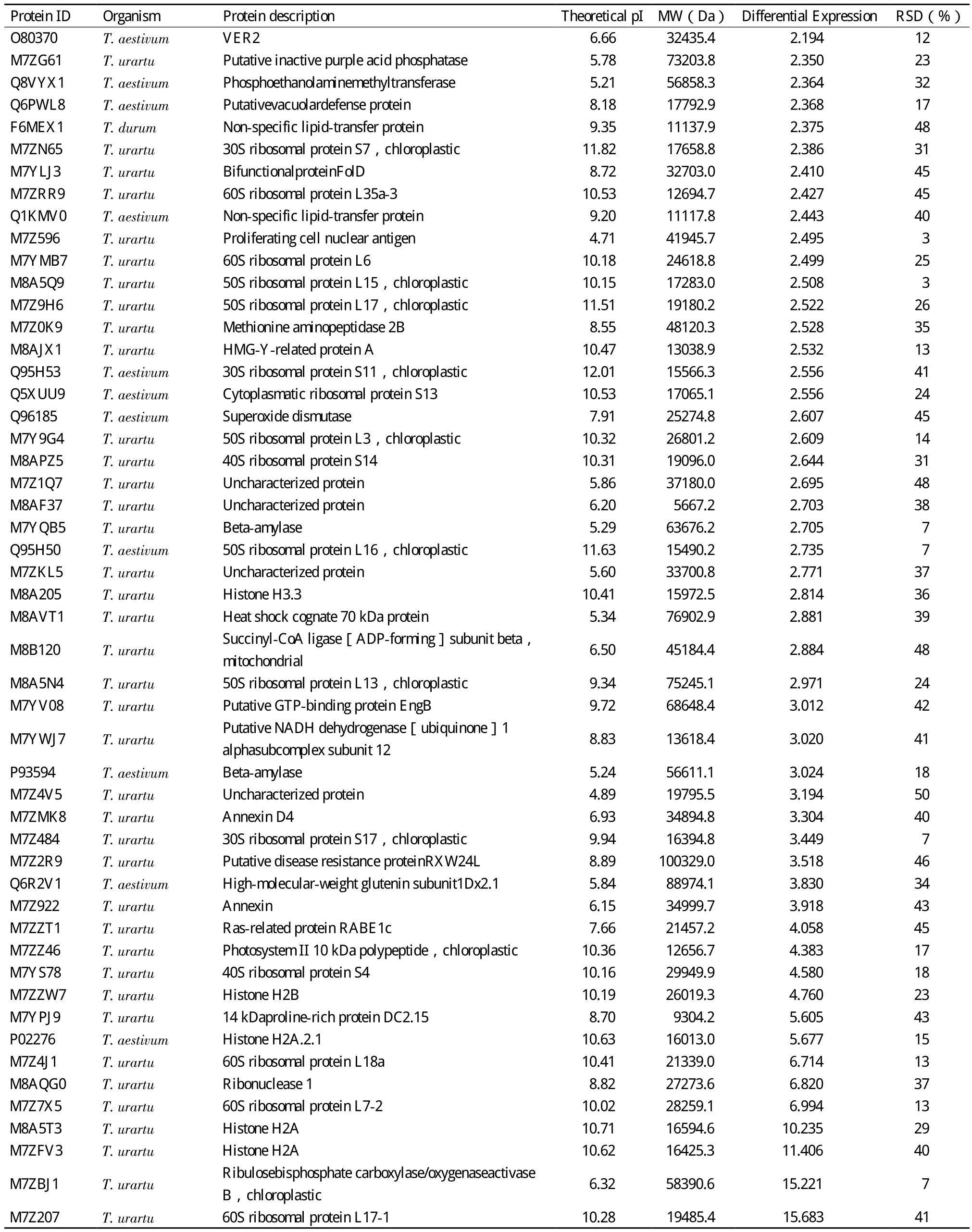
续表
按照UNIPROT(http://www.uniprot.org/)数据库中的各蛋白质的GO注释分别进行细胞成分、分子功能和生物学途径分类。结果(图2)显示,102个差异蛋白中有63个没有明确的亚细胞定位,其余主要来源于细胞基质(36.3%)与核糖体(23.6%)等细胞器;分子功能涉及结合(41.2%)、催化(37.3%)等方面;生物学途径GO分析表明,这些蛋白主要参与代谢(58.8%)、细胞过程(50.0%)、应激(3.9%)等生物学进程。这些蛋白中,具有细胞成分(Cellular component)GO注释信息的蛋白39个,占总差异蛋白的38.2%,其中细胞基质蛋白37个,基质蛋白中也分布于核糖体、细胞核和膜系统的蛋白分别为24、8和3个,另有2个膜蛋白不分布于细胞基质;具有分子功能(Molecular function)GO注释信息的蛋白85个,占到总差异蛋白的84.3%,其中具有结合功能的蛋白42个,在这42个蛋白中也具有催化活性、结构大分子功能的蛋白分别为21个和5个,具有催化活性的蛋白一共是38个,其中1个同时具有大分子结构功能,另有7个具有其他分子功能的蛋白;具有生物学途径(Biological process)GO注释信息的蛋白75个,占总差异蛋白的73.6%,其中代谢途径相关蛋白60个,在这60个蛋白中同时与细胞进程、应激反应、转运过程以及生物调节过程相关的蛋白分别有41、2、1和5个。与细胞进程相关的蛋白一共有51个,其中同时与转运以及生物调节过程相关的蛋白分别为3个和6个。应激反应、转运过程和生物调节过程相关的蛋白总数分别为4、 6和7个。

图2 T29与TYr10叶片间差异蛋白的GO注释图
3 讨论
样品制备是质谱非标记定量分析的关键步骤之一,其成功与否对研究结果影响巨大,但目前还没有一种提取植物叶片完整蛋白质的标准方法[19]。由于小麦等植物叶片中含有RuBisCo等一些高拷贝数的持家蛋白,可能导致某些调控因子和信号因子等低丰度蛋白难以检测,对样品进行去除高丰度蛋白处理可以在一定程度上缓解此类问题,然而此类方法成本昂贵或操作步骤复杂,容易造成蛋白的丢失[20,21]。本研究运用TCA/丙酮沉淀法对小麦叶片总蛋白进行提取,蛋白提取率分别为0.58%和0.60%,达到目前的一般标准[9,22],在此基础上首次运用质谱非标定量法对TYr10与T29进行了比较蛋白质组学研究。结果显示,在两个样品中共鉴定到2 257个蛋白,其中具有准确定量信息的蛋白1 549个,从已定量的蛋白中共筛选到102个差异表达蛋白。
由于在不同真核生物中的大多数基因拥有相似的生物学功能,因此在某些物种获得基因或蛋白质的生物学信息可辅助揭示其他物种中相应基因或蛋白质的功能,综合这些信息的GO注释目前已成为功能基因生物信息学分析的核心内容之一[23,24]。本研究从GO数据库中获取了102个差异表达蛋白所属的细胞成分、生物学途径和分子功能的GO注释,并人工对差异蛋白进行GO功能分析和图示化。这一结果表明该鉴定蛋白质组数据具有较好的生物学功能覆盖范围,包括光合作用、糖代谢以及能量代谢等一系列生物过程,这与报导过的植物抗病相关
研究结果相一致[25-27]。
在已鉴定的差异蛋白中,代谢相关蛋白60个,其中Q96185为超氧化物歧化酶(Superoxide dismutase,SOD)。SOD广泛存在于真核细胞与原核细胞的细胞质、线粒体和叶绿体中,可清除生物体内超氧阴离子自由基,有效地抵御氧自由基对有机体的伤害[28],研究还发现大豆在感染SMV后引起了活性氧的累积,诱导防御酶活性的增强,从而使SOD活性的上升[29],而SOD在TYr10中表达有明显上升可能与其抗病性有关。M7Z0K9为甲硫氨酰氨肽酶(Methionine aminopeptidase,MetAP)。MetAP在细胞内的主要功能是切除细胞内新合成蛋白的N端甲硫氨酸,在细胞信号转导、蛋白互作、肿瘤生长以及细菌和病毒感染等生理病理过程发挥着重要的作用[30,31],但是与TYr10的抗病性的关系尚不清楚。M7ZME5为溶酶体-β-葡萄糖苷酶(Lysosomal beta glucosidase)。马成等[8]曾报道Taichung29*6/Yr5叶片在接种条锈菌CYR32后β-葡萄糖苷酶水平下降,本研究发现溶酶体-β-葡萄糖苷酶在未接种的TYr10中表达较T29低,提示该蛋白可能与抗病性有一定相关性。本研究还鉴定到6个与运转相关的差异蛋白,其中Q5G1L6为铁蛋白(Ferritin)。铁蛋白广泛存在于动物、植物和微生物体内,在植物中作为专门的贮铁蛋白,是植物光合作用和固氮等生化反应的铁源,不仅调节体内铁的含量,而且是一种重要的胁迫反应蛋白,在植物的发育、抵抗氧化损害等方面发挥重要的作用[32],该蛋白在TYr10中的高表达可能与其抗病性有关。此外,还鉴定到4个应激反应相关的差异蛋白,其中M7Z2R9(Putative disease resistance protein RXW24L)与Q6PWL8(Putative vacuolar defense protein)在TYr10表达明显提高,可能与该近等基因系的抗病机制相关,值得进一步研究。多年来,育种工作者一直以选育和推广抗病品种作为病害的主要防治手段,且颇见成效,但小麦抗条锈性丧失和抗条锈病机理一直是未能解决的问题。对近等基因系Taichung 29*6/Yr10差异表达蛋白的分析,为理解小麦抗条锈性丧失和研究Yr10基因的抗条锈病机理提供了新的线索和思路。
4 结论
利用质谱非标定量技术可有效地进行小麦蛋白质组定量分析,运用该技术从小麦抗病单基因近等基因系Taichung 29*6/Yr10和背景品种Taichung 29叶片中共鉴定到102个差异表达蛋白。已鉴定到的差异表达蛋白大多定位于细胞基质以及核糖体等细胞器,具备结合、催化等功能,主要参与代谢、细胞过程、应激等生物学进程,其中超氧化物歧化酶、甲硫氨酰氨肽酶、溶酶体-β-葡萄糖苷酶和铁蛋白等可能与小麦抗病单基因近等基因系Taichung 29*6/Yr10的抗病性有一定相关性。
致谢:质谱分析和数据处理得到赛默飞世尔科技(中国)有限公司色谱与质谱部聂爱英博士的帮助。
[1] Wan A, Zhao Z, Chen X, et al. Wheat stripe rust epidemic and virulence ofPuccinia striiformisf. sp.triticiin China in 2002[J]. Plant Disease, 2004, 88(8):896-904.
[2] Wan A, Chen X, He Z. Wheat stripe rust in China[J]. Crop and Pasture Science, 2007, 58(6):605-619.
[3] Wellings CR. Global status of stripe rust:a review of historical and current threats[J]. Euphytica, 2011, 179(1):129-141.
[4] Zeng SM, Luo Y. Long-distance spread and interregional epidemics of wheat stripe rust in China[J]. Plant Disease, 2006, 90(8):980-988.
[5] Wan A, Chen X. Virulence, frequency, and distribution of races ofPuccinia striiformisf. sp.triticiandP.striiformisf. sp.hordeiidentified in the United States in 2008 and 2009[J]. Plant Disease, 2012, 96(1):67-74.
[6] Cox J, Mann M. Is proteomics the new genomics?[J]. Cell, 2007, 130(3):395-398.
[7] Rampitsch C, Bykova NV, McCallum B, et al. Analysis of the wheat andPuccinia triticina(leaf rust)proteomes during a susceptible host-pathogen interaction[J]. Proteomics, 2006, 6(6):1897-1907.
[8] 马成, 徐世昌, 徐琴, 等.抗条锈病小麦品系Taichuang29*6/ Yr5接种条锈菌CY32后的蛋白质组学分析[J].中国农业科学, 2009, 42(5):1616-1623.
[9] 胡新明, 杨友才, 潘映红.小麦叶片胞间洗脱液的蛋白质组学
检测分析[J].生物技术通报, 2011(8):91-98.
[10] Jorrín-Novo JV, Maldonado AM, Echevarría-Zomeño S, et al. Plant proteomics update(2007-2008):second-generation proteomic techniques, an appropriate experimental design, and data analysis to fulfill MIAPE standards, increase plant proteome coverage and expand biological knowledge[J]. Journal of Proteomics, 2009, 72(3):285-314.
[11] Ross PL, Huang YN, Marchese JN, et al. Multiplexed protein quantitation inSaccharomyces cerevisiaeusing amine-reactive isobaric tagging reagents[J]. Molecular & Cellular Proteomics, 2004, 3(12):1154-1169.
[12] Gygi SP, Rist B, Gerber SA, et al. Quantitative analysis of complex protein mixtures using isotope-coded affinity tags[J]. Nature Biotechnology, 1999, 17(10):994-999.
[13] Kang UB, Yeom J, Kim H, et al. Quantitative analysis of mTRAQ-labeled proteome using full MS scans[J]. Journal of Proteome research, 2010, 9(7):3750-3758.
[14] Erba EB, Bergatto E, Cabodi S, et al. Systematic analysis of the epidermal growth factor receptor by mass spectrometry reveals stimulation-dependent multisite phosphorylation[J]. Molecular & Cellular Proteomics, 2005, 4(8):1107-1121.
[15] Wang G, Wu W, Pisitkun T, et al. Automated quantification tool for high-throughput proteomics using stable isotope labeling and LCMSn[J]. Anal Chem, 2006, 78(16):5752-5761.
[16] Zhang K, Chen Y, Zhang Z, et al. Identification and verification of lysine propionylation and butyrylation in yeast core histones using PTMap software[J]. Journal of Proteome Research, 2008, 8(2):900-906.
[17] 刘伟霞, 潘映红.适用于小麦叶片蛋白质组分析的样品制备方法[J].中国农业科学, 2007, 40(10):2169-2176.
[18] Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of proteindye binding[J]. Analytical Biochemistry, 1976, 72(1):248-254.
[19] Bodzon-Kulakowska A, Bierczynska-Krzysik A, Dylag T, et al. Methods for samples preparation in proteomic research[J]. Journal of Chromatography B, 2007, 849(1):1-31.
[20] Kim ST, Cho KS, Jang YS, et al. Two-dimensional electrophoretic analysis of rice proteins by polyethylene glycol fractionation for protein arrays[J]. Electrophoresis, 2001, 22(10):2103-2109.
[21] Cellar NA, Kuppannan K, Langhorst ML, et al. Cross species applicability of abundant protein depletion columns for ribulose-1, 5-bisphosphate carboxylase/oxygenase[J]. Journal of Chromatography B, 2008, 861(1):29-39.
[22] 刘静, 潘映红, 徐琴, 等.小麦叶片蛋白质组的2D-LC分离及Nano LC-MS/MS分析[J].中国农业科学, 2009, 42(3):772-780.
[23] Matis M, Žakelj-Mavrič M, Peter-Katalinić J. Mass spectrometry and database search in the analysis of proteins from the fungusPleurotus ostreatus[J]. Proteomics, 2005, 5(1):67-75.
[24] Wu X, Zhu L, Guo J, et al. Prediction of yeast protein-protein interaction network:insights from the Gene Ontology and annotations[J]. Nucleic Acids Research, 2006, 34(7):2137-2150.
[25] Li Y, Zhang Z, Nie Y, et al. Proteomic analysis of salicylic acidinduced resistance toMagnaporthe oryzaein susceptible and resistant rice[J]. Proteomics, 2012, 12(14):2340-2354.
[26] Bernardo L, Prinsi B, Negri AS, et al. Proteomic characterization of the Rph15 barley resistance gene-mediated defence responses to leaf rust[J]. BMC Genomics, 2012, 13(1):1-16.
[27] Zhao F, Fang W, Xie D, et al. Proteomic identification of differentially expressed proteins inGossypium thurberiinoculated with cottonVerticillium dahliae[J]. Plant Science, 2012, 185:176-184.
[28] 陈鸿鹏, 谭晓风.超氧化物歧化酶(SOD)研究综述[J]. 经济林研究, 2007, 25(1):59-65.
[29] 栾晓燕, 陈怡, 杜维广, 等.不同抗性大豆品种感染SMV后过氧化物酶, 多酚氧化酶, 超氧化物歧化酶的变化分析[J].大豆科学, 2001, 20(3):200-203.
[30] Chang SY, McGARY EC, Chang S. Methionine aminopeptidase gene of Escherichia coli is essential for cell growth[J]. Journal of Bacteriology, 1989, 171(7):4071-4072.
[31] Cutforth T, Gaul U. A methionine aminopeptidase and putative regulator of translation initiation is required for cell growth and patterning inDrosophila[J]. Mechanisms of Development, 1999, 82(1-2):23-28.
[32] Andrews SC, Harrison PM, Yewdall SJ, et al. Structure, function, and evolution of ferritins[J]. Journal of Inorganic Biochemistry, 1992, 47(1):161-174.
(责任编辑 马鑫)
Label-free Quantitative Proteomics Analysis of a Wheat Near-isogenic Line Pair with Q Exactive Mass Spectrometer
Huang Yu1,2,3Feng Jing4Rao Liqun1Xu Shichang4Pan Yinghong2,3
(1. College of Bbioscience and Biotechnology,Hunan Agriculture University,Changsha 410128;2. Institute of Crop Science,Chinese Academy of Agricultural Sciences,Beijing 100081;3. The National Key Facility for Crop Gene Resources and Genetic Improvement,Beijing 100081;4. Institute of Plant Protection,Chinese Academy of Agricultural Sciences,Beijing 100193)
In this study, the protein expression of a wheat near-isogenic line pair, Taichung 29*6/Yr10 and Taichung 29, have been compared with label-free quantitative proteomic approach based on liquid chromatography tandem Q Exactive hybrid quadrupole-Orbitrap mass spectrometer. By MASCOT database search, a total of 2 257 proteins were identified from both of the samples, and among of them 1 549 proteins were accurately quantitated and 102 differentially expressed proteins(>2 fold)were detected. Gene ontology(GO)analysis showed that these differential proteins were mainly localized to cell matrix and organelles such as ribosome, and mainly involved in binding and catalytic functions. Among of 102 proteins which mainly take part in metabolic, cellular and stress responses processes, Superoxide dismutase, Methionine aminopeptidase, Lysosomal-β-glucosidase, and Ferritin may play some important roles in Taichung 29*6/Yr10 resistance to Yellow rust.
Wheat Yellow rust Resistance Proteomics Label-free quantitation Q Exactive Mass Spectrometer
2013-12-16
国家“863”高技术研究发展计划(2008AA10Z115),国家自然科学基金项目(30971874),转基因生物新品种培育科技重大专项(2009ZX08012-011B)
黄宇,男,硕士研究生,研究方向:生物化学与分子生物学;E-mail:nictkk@163.com
潘映红,男,研究员,研究方向:生物化学与分子生物学;E-mail:panyinghong@caas.cn
