鸭茅品种的SCoT遗传变异分析
2014-01-02蒋林峰张新全黄琳凯马啸严德飞胡强付玉凤
蒋林峰,张新全 ,黄琳凯,马啸,严德飞,胡强,付玉凤
(1.四川农业大学动物科技学院草业科学系,四川雅安625014;2.重庆市垫江县畜牧生产站,重庆408300)
目标起始密码子多态性(start codon targeted polymorphism,SCoT),是基于SRAP(sequence-related amplified polymorphism,相关扩增多态性)[1]的一种新型目的基因分子标记技术,其产生的显性多态性偏向候选功能基因区[2]。主要依据植物基因中ATG翻译起始位点侧翼序列的保守性而设计[3-4]。另外,较高退火温度的设计有助于减少假阳性扩增的可能性[5]。与其他标记相比,SCoT标记结合了ISSR(inter simple sequence repeat,简单重复序列间区)[6-7]和RAPD(randomly amplified polymorphic DNA,随机扩增多态性DNA)的优点,具引物通用性,稳定性好,重复性好等优点,同时能有效产生相关性状多态,更好地反映物种遗传多样性和亲缘关系[8]。自2009年开发以来,已有其用于葡萄(Vitis vinifera)[9]、芒果(Mangifera indica)[10]、花生(Arachis hypogaea)[11]、土豆(Solanum tuberosum)[12]等农作物和植物的相关研究报道。但就牧草研究领域,目前仅有SCoT标记用于苜蓿(Medicago sativa)[4]种质遗传分析的少量报道。
鸭茅(Dactylis glomerata)为禾本科(Poaceae)早熟禾亚科(Pooideae)鸭茅属(Dactylis)多年生疏丛型禾草[13-14],是鸭茅亚种中重要的同源四倍体栽培种[15],起源于欧洲、北非和亚洲温带地区[16],因草质柔嫩、叶量丰富、耐荫性强、适口性好等优点,在世界各地被广泛应用于饲草栽培和干草制作[17],是温带和亚热带地区重要优良牧草。我国是鸭茅起源地之一,已发现野生鸭茅生长地约26个[18],可分为二倍体(2n=2x=14)、四倍体(2n=4x=28)及稀有的六倍体(2n=6x=42)3种类型[19],近年来,鸭茅大量应用于我国草地畜牧业及生态建设,取得了良好的经济和生态效益。
到目前,我国审定登记鸭茅品种仅为8个,且只有“宝兴”、“川东”、“古蔺”3个品种通过野生栽培驯化而来,其余5个皆为引进品种,国内鸭茅品种遗传基础相对狭窄,引进品种普遍表现出在当地抗性低、适应性差等缺点,难以满足我国生态治理和畜牧业发展的需求。采用不同的育种方法,综合选育利用我国丰富的鸭茅种质资源已经刻不容缓。而纵观全球,国外通过不同的选育目标和育种方式已获得近500个鸭茅品种,品种适应性强。采用不同的技术手段开展鸭茅种质遗传研究,对于鸭茅育种具有重要意义。同时,国内外学者对鸭茅的研究已从形态学[20]、细胞学[21]、同工酶[22]深入到分子水平。分子标记因不受环境影响、效率快、分辨率高等优点被广泛应用。谢文刚等[23]利用SSR(simple sequence repeat,简单重复序列)对11份鸭茅种质的110个单株遗传分析表明,我国西南地区鸭茅种质遗传多样性丰富,其遗传变异主要存在于种质内和地理区域内;Peng等[24]利用AFLP(amplified fragment length polymorphism,扩增片段长度多态性)对32份鸭茅分析表明,鸭茅遗传多样性与染色体倍性、地理分布等显著相关;万刚等[25]对23份鸭茅二倍体、四倍体SSR研究表明,四倍体较二倍体遗传多样性更为丰富,是鸭茅品种选育的优良材料。这些研究主要集中于鸭茅野生材料、栽培驯化品种等的研究,但对于鸭茅国内栽培驯化品种与引进品种的遗传变异对比研究,国内外均未有报道。了解我国现有鸭茅栽培驯化品种的遗传背景和变异,有助于保证我国鸭茅种质资源的合理利用和丰富我国鸭茅野生驯化栽培驯化品种的遗传多样性。
本研究首次利用SCoT标记技术对来自世界4大洲的32个鸭茅品种进行遗传变异分析,比较栽培驯化品种与引进品种之间的遗传差异,以期为进一步加快我国鸭茅育种进程和针对性提高鸭茅品种生态适应能力及利用价值等提供理论依据。
1 材料与方法
1.1 试验材料
供试材料为32个鸭茅品种,包括8个国内栽培驯化品种(系)和24个国外引进品种,均为四川农业大学草业科学系从国内及国外采集和收集而来(表1),于2012年9月种植于四川农业大学雅安草业科学试验基地。
1.2 方法
1.2.1 DNA提取 每份种质随机选取25个单株的幼嫩叶片等量混合,采用Doyle等[26]的CTAB(cetyl trimethyl ammonium bromide,十六烷基三甲基溴化铵)法提取DNA,并用1%琼脂糖凝胶电泳和紫外分光光度计分别检测其DNA的纯度和浓度,合格的样品保存于-20℃低温冰箱,实验开始时,取出部分按照其测得浓度稀释至10 ng/μL,4℃冰箱保存备用。
1.2.2 SCoT引物筛选 所用80个SCoT引物由澳大利亚Collard和Mackill[3](SCoT1~SCoT36)及中国广西农业大学Luo等[5](SCoT37~SCoT80)提供,引物由上海生工生物技术有限公司合成。PCR所用Mix混合液(含有10×PCR buffer、Mg2+、dNTPs)和Taq酶均购自天根科技生化公司。采用田间形态差异较大的4个品种,即栽培驯化品种“宝兴”、引进品种“安巴”、“Chinook”和新品系“YA01472”,对引物进行预先筛选,从中选取扩增条带清晰且重复性较好的22个SCoT引物,用于供试32个鸭茅品种的进一步PCR扩增(表2)。
1.2.3 SCoT-PCR扩增 本实验PCR程序参考植物水稻(Oryza sativa)[3]和牧草苜蓿[4]并有所改动,扩增体系优化为15 μL:包括DNA 模板1 μL(10 ng/μL),引物1.5 μL(10 pmol/μL),Mix混合液7.5 μL,Taq酶0.4 μL(2.5 U/μL),ddH2O 4.6 μL。PCR 反应程序为94℃预变性3 min;94℃变性50 s,50℃退火1 min,72℃延伸2 min,36个循环;72℃延伸5 min,最后冷却至4℃保存。PCR扩增产物在含0.05μL/mL Gelred(10000×水溶液)的1.8%琼脂糖凝胶中电泳,待溴酚蓝电泳至凝胶尾端约1 cm时停止电泳,约需2.5~3.0 h。电泳完毕观察,再用凝胶成像系统拍照。
1.2.4 数据分析 SCoT是显性标记,对获得的清晰可重复的DNA条带进行统计,在相同迁移位置上将稳定出现的条带的有或无赋值为1和0进行统计,构成原始数据矩阵。利用软件Excel 2007和POPGENE 1.31[27]计算多态性条带百分比(percentage of polymorphic bands,PPB),Nei氏遗传多样性指数(Nei’s gene diversity,H)[28]和Shannon信息多样性指数(Shannon’s information index of diversity,I)。利用WINAMOVA 1.55对国内栽培驯化品种与引进品种的遗传变异进行分子方差分析(analysis ofmolecular variance,AMOVA)[29]。POPGENE和AMOVA数据输入文件均由软件 DCFA1.1[30]生成。利用软件 FreeTree[31]基于 Nei-Li[32]遗传相似系数(genetic similarity,GS)的不加权成对群算术平均法(unweighted pair-group method with arithmetic averages,UPGMA)计算各品种的遗传距离(genetic distance,GD),按公式GD=1-GS计算。采用Dendroscope 3[33]构建各品种的聚类树,利用NTsyspc V2.1[34]进行鸭茅品种的主成分分析(principal coordinate analysis,PCoA)。
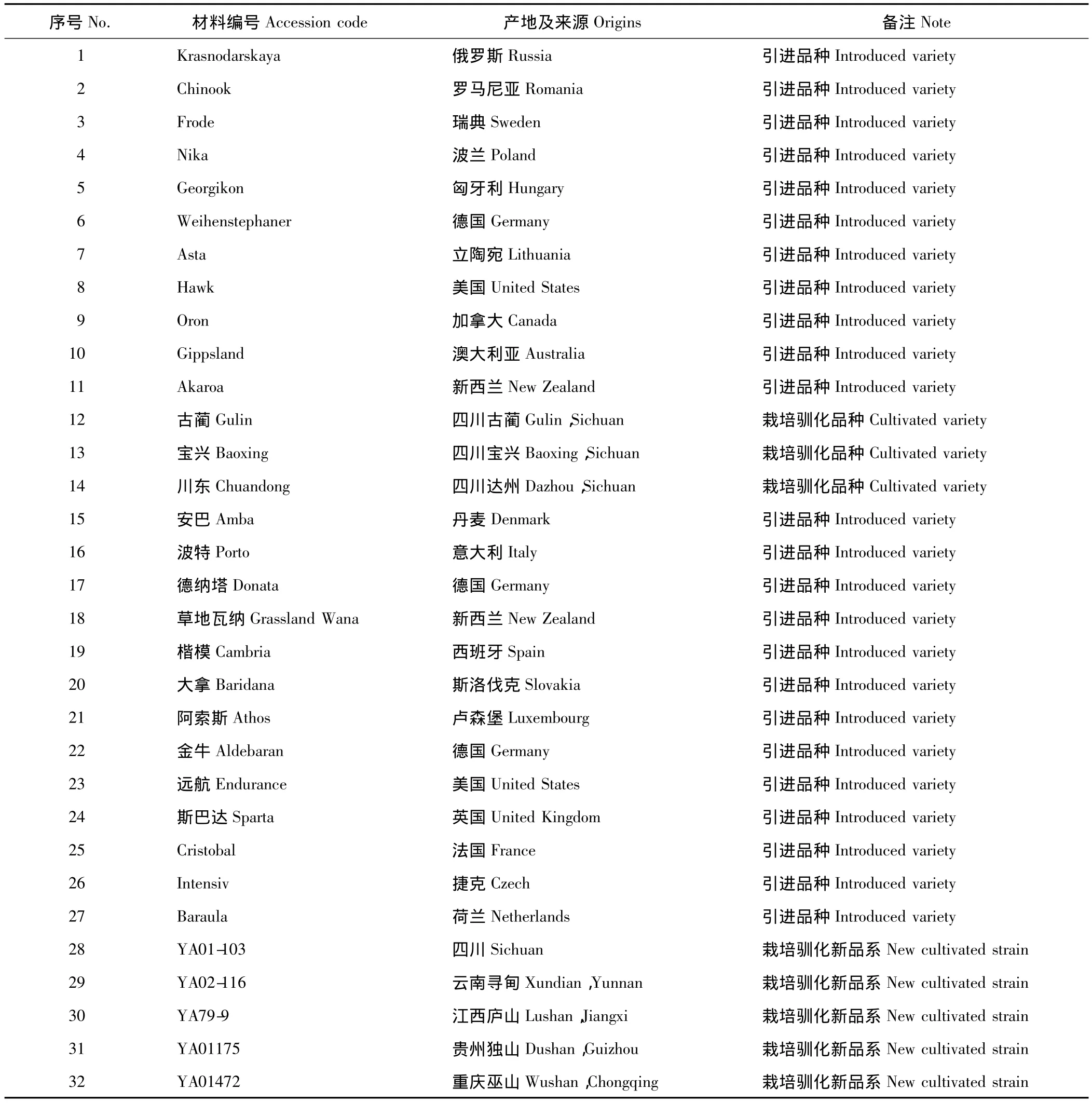
表1 供试鸭茅材料名称及来源Table 1 Names and sources of D.glomerata used in this study
2 结果与分析
2.1 SCoT标记多态性分析
22个引物共扩增出308条清晰可辨条带,扩增片段大小在250~2000 bp,大部分集中在250~1500 bp(图1),平均每个引物扩增条带数为14条,条带变化为9条(SCoT8,SCoT9和SCoT20)到19条(SCoT40),其中,多态性谱带245条,平均每个引物扩增多态性条带数为11.14条,多态性比率(PPB)为79.55%,多态性条带变化为5条(SCoT9,SCoT11和SCoT12)到18条(SCoT65),表明SCoT标记能检测到较多的遗传位点,获得多态性较好的PCR结果(表3)。
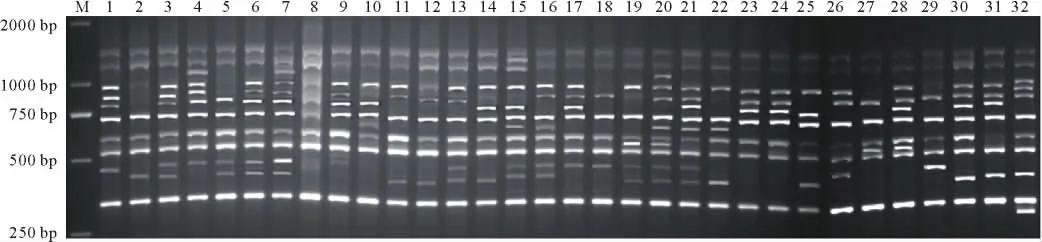
图1 引物SCoT 23对鸭茅品种的扩增结果Fig.1 Am plified resultsw ith primer No.SCoT 23 on orchardgrass varieties
2.2 供试鸭茅品种遗传距离(GD)分析
根据0,1原始数据表征矩阵,利用软件Freetree[31]基于Nei-Li遗传相似系数得到供试鸭茅品种的遗传距离,用于分析其相互之间的亲缘关系。32份鸭茅品种遗传距离范围为0.0251~0.3157,平均遗传距离为0.1916,其中来自法国的Cristobal和来自捷克的Intensiv遗传距离最大,表明其之间的亲缘关系最远,来自匈牙利的Georgikon和来自波兰的Nika遗传距离最小,表明其亲缘关系最近。总体上遗传距离的差异呈现了不同供试鸭茅品种来源地的分布差异,品种间遗传差异较大。将所有品种按照育种背景划分为国内栽培驯化品种和国外引进品种,可以进一步分析2种选育背景下不同鸭茅品种之间的遗传分化和差异,结果发现栽培驯化品种内部的遗传距离范围为0.0601~0.1802,平均遗传距离为0.1242,其中“宝兴”和“川东”遗传距离最小,其都为国审栽培驯化品种,“YA01472”和“YA02-116”遗传距离最大,其为鸭茅栽培驯化新品系,表明目前国内登记的鸭茅栽培驯化品种间遗传相似度较高,新品系“YA01472”和“YA02-116”遗传多样性较已经登记的栽培驯化品种(“宝兴”、“古蔺”、“川东”)有所提高;而引进品种内部的遗传距离范围为0.0251~0.3157,平均遗传距离为0.1952,可见国内栽培驯化品种的遗传多样性水平低于引进品种。
2.3 鸭茅类群的遗传变异比较分析
将32份鸭茅品种划分为栽培驯化品种类群和国外引进品种类群,比较他们之间的遗传变异。采用软件AMOVA 1.55[29]分析其类群之间和之内的方差、方差分量及贡献率(表 4),采用 POPGENE version 1.32[27]分析其类群的遗传多样性和反应其类群的等位基因丰富度和均匀程度(表5)。分子方差分析(AMOVA)结果表明,16.32%的遗传变异存在于两类群之间,两类群之内的遗传变异较高,占有83.68%,2种品种类群之间和之内的差异均极显著(P<0.001)。此外,供试鸭茅品种类群间的表型预测(Фst)值为0.1630,也说明遗传结构的变异主要存在于品种类群之内。由表5可知,供试鸭茅品种基因多样性指数为0.2646,Shannon指数为0.3983。其中引进品种的多态性条带(NPB),多态性比率(PPB),基因多样性指数(H),Shannon指数(I)都高于栽培驯化品种,基本代表了鸭茅物种的整体遗传水平,与2.2中分析的遗传距离分析结果相符合,说明鸭茅引进品种较目前国内普遍存在的栽培驯化品种有更为丰富的遗传多样性和变异水平,这可能是国外品种选育方法和选育材料较为多元化的结果。
2.4 基于Nei-Li遗传相似系数的聚类分析
利用Freetree[31]软件计算出供试各鸭茅品种间的Nei氏遗传一致度和遗传距离的无偏估计值,然后用Dendroscope 3[33]绘制 UPGMA 聚类图。总体看来,供试32份鸭茅品种主要可分为6类(图2)。类群I有8个品种(系),包括来自中国的3个栽培驯化品种(“宝兴”、“古蔺”、“川东”)和 5个野生驯化新品系(“YA79-9”、“YA01-103”、“YA02-116”、“YA01175”、“YA01472”);类群II包括3个品种,即来自大洋洲的“Gippsland”、“草地瓦纳”和“Akaroa”;类群 VI包括 8个品种,即来自欧洲的“Georgikon”、“Nika”、“Weihenstephaner”、“金牛”、“德纳塔”、“大拿”、“Asta”和“Intensiv”;类群 III包括来自南美洲的 3个品种(“Hawk”、“远航”、“Oron”)及来自欧洲的 6 个品种(“Krasnodarskaya”、“Frode”、“安巴”、“波特”、“斯巴达”、“阿索斯”);其余2个类群(IV和V)包括了来自欧洲的其余供试鸭茅品种。由聚类分析可知,目前国内已经登记的鸭茅栽培驯化品种间具有一定的遗传相似度,遗传距离较近,3个品种(“古蔺”、“宝兴”、“川东”)育成登记时间分别为1995,1999和2003年,可能由当时较为简单的选育方法、相似的生态地理环境等原因导致,而新品系“YA02-116”较之前栽培驯化品种有所改善,但遗传变异仍较为狭窄,这与前面的分析结果一致,可能是由于不同生境材料混合采样及选育过程中混合选择等原因;而反观国外选育的品种,其大多数为综合品种,其同一来源的品种间存在较大的遗传距离和遗传差异,但总体上供试品种的聚类与地理分布存在一定的相关性,且其品种继承了更为丰富的遗传多样性和更为一致的群体一致性,利于品种的推广和应用。
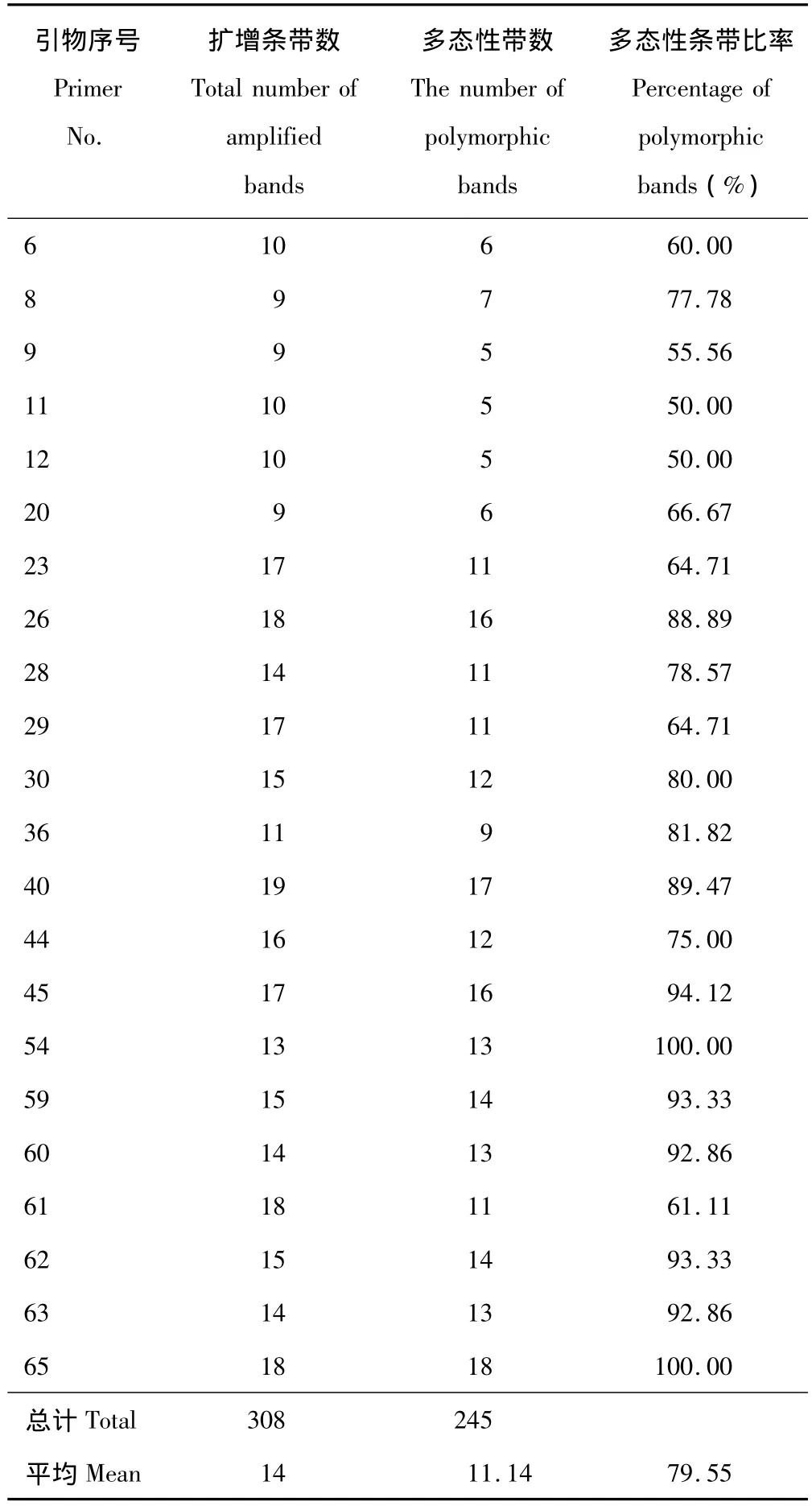
表3 SCoT标记扩增结果Table 3 Amp lified results of SCoT markers
2.5 主成分分析
主成分分析(PCoA)可作为种质聚类分析结果的验证和对比,同时各种质在主成分分析二维或三维图上的远近关系可以更为直观的反映它们之间的亲缘关系和遗传差异。利用原始矩阵数据基于遗传相似系数(GS),用NTsys-pc V2.1[34]软件对供试32份鸭茅品种进行主成分分析,前3个主成分所能解释的遗传变异为46.08%,并根据第一、第三主成分进行作图(图3)。主成分结果与聚类分析结果基本一致,整体上显示出国内的栽培驯化品种(系),尤其是3个国审鸭茅栽培驯化品种间亲缘关系较近,遗传丰富度和遗传变异水平较引进品种稍差,通过栽培驯化品种和引进品种遗传变异的对比,反思目前国内普遍使用的选育手段是否具有代表性、品种遗传一致度是否较好等问题,对于今后我国的鸭茅新品种选育工作和加快鸭茅育种进程具有一定的指导意义。

表4 鸭茅品种分子变异方差分析(AMOVA)Table 4 Analysis ofmolecular variance(AMOVA)for variety of orchardgrass
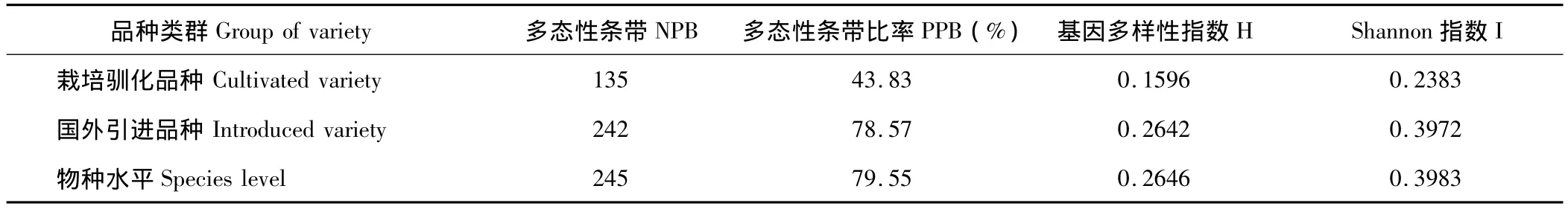
表5 2种品种类群遗传多样性指数Table 5 Genetic diversity index of two varieties groups
3 讨论
3.1 SCoT标记的多态性
SCoT标记是一种基于翻译起始位点(translation initiation site,TIS)的目标分子标记新技术,具有操作简单、引物通用性、多态性高、成本低、重复性好等优点[2],其较多的基因组基因形成了较多的引物结合位点,且其单引物的设计,使得那些较近而又反向结合的引物结合位点之间的片段得以有效扩增。扩增不确定性介于外显子与内含子之间,大幅度地提升了其引物的多态性[8]。较其他分子标记(AFLP,ISSR,SSR,SRAP,RAPD等),其与功能基因相关,更能反应相关功能性状多态[11],是用于农作物、水果和牧草等植物种质资源鉴定、保护和利用,高密度遗传连锁图谱构建,分子标记辅助育种,基因定位和克隆等研究的非常有效的一种分子标记。
本研究首次将SCoT标记技术应用于鸭茅品种的DNA多态性鉴定,其能在鸭茅品种资源中检测出较丰富的遗传多态性,这与鸭茅品种资源间具有较丰富的表型性状多态性相一致。扩增平均多态性条带为11.14条,而谢文刚等[23]使用SSR标记对来自中国西南地区鸭茅的遗传多样性研究多态性条带为6.8条,Guo等[9]采用SCoT标记对64份葡萄品种研究的多态性条带为7.7条,可见SCoT标记能够产生较多的多态位点用于鸭茅遗传变异研究。另外,其多态性位点率为79.55%,而Luo等[5]采用SCoT标记对来自中国的50份芒果种质研究的多态性为76.19%,Luo等[35]采用ISSR标记对23份芒果种质分析的多态性为55.77%,熊发前等[8]对花生属采用与功能基因相关的SCoT研究多态性为31.80%,韩国辉等[2]对柑橘(Citrus)SCoT研究多态性为54.80%,由此可知,SCoT标记可以作为研究鸭茅遗传多样性的有效手段,其得到的分子标记极有可能是功能基因的一部分,是进一步开发特定功能基因标记和建立分子标记辅助育种技术体系的基础。
3.2 鸭茅栽培驯化品种与引进品种遗传差异
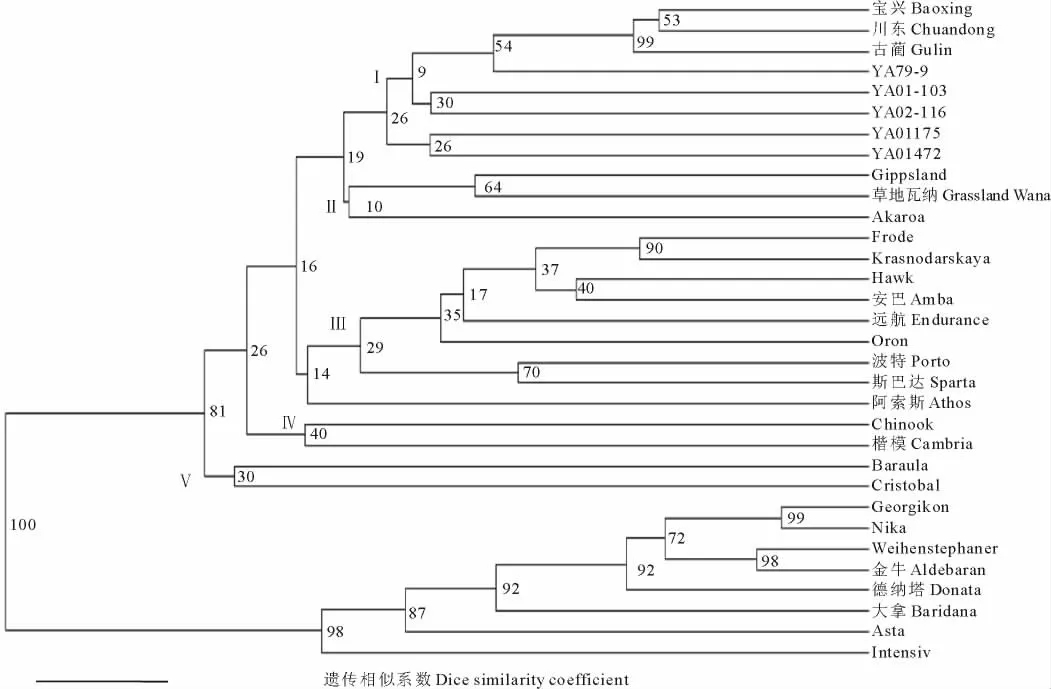
图2 SCoT标记对32份鸭茅品种亲缘关系聚类图Fig.2 Dendrogram of the relationship UPGMA cluster of 32 orchardgrass varieties based on SCoT markers
本研究采用SCoT标记技术开展鸭茅栽培驯化品种与引进品种的遗传变异比较,研究表明,国内鸭茅栽培驯化品种平均遗传距离为0.1242,多态性条带为135条,基因多样性指数为0.1596,Shannon指数为0.2383,而鸭茅引进品种的遗传多样性从以上几个水平都远高于栽培驯化品种,基本代表了鸭茅物种的遗传变异水平。从侧面揭示了目前国内鸭茅品种遗传基础狭窄的现状。通过分子方差分析表明,鸭茅栽培驯化品种和引进品种2个类群的遗传变异主要还是分布于类群内部,尤其是引进品种内部的不同鸭茅品种间具有更为丰富的遗传差异。聚类分析和主成分分析的结果基本一致,供试32个鸭茅品种主要可分为6类,其中来自国内的8个栽培驯化品种聚为类群I,进一步印证了目前国内鸭茅栽培驯化品种内部遗传变异较小的瓶颈。这与万刚等[25]的研究结果一致,认为3个国内栽培驯化品种(“宝兴”、“川东”、“古蔺”)间的遗传变异较为狭窄,可能是缘于3个鸭茅栽培驯化品种采集地相似的自然气候地理条件和较为传统的育种方式等原因。
总的来说,就鸭茅栽培驯化品种和引进品种的遗传分化而言,引进品种和国内栽培驯化品种间差异较大,引进品种的遗传多样性更为丰富,而栽培驯化品种相对来说整齐度差,品种结实性差,适应范围小。究其原因,可能是国外鸭茅育种家在育种方法和育种材料的选择上,较国内科研单位更为先进和合理,其针对性强,工作延续性强,选育时间长,品种性状更为稳定。目前采用较多的是选择在各方面性状表现较为优良的不同材料,让其自由传粉,经过多代选择和淘汰,并采取一定的育种手段,使优良基因得到稳定遗传,这样选育得到的品种具有较为丰富的遗传多样性和较大的生境适应能力,及更为稳定的群体一致性。
3.3 鸭茅新品种选育及其利用
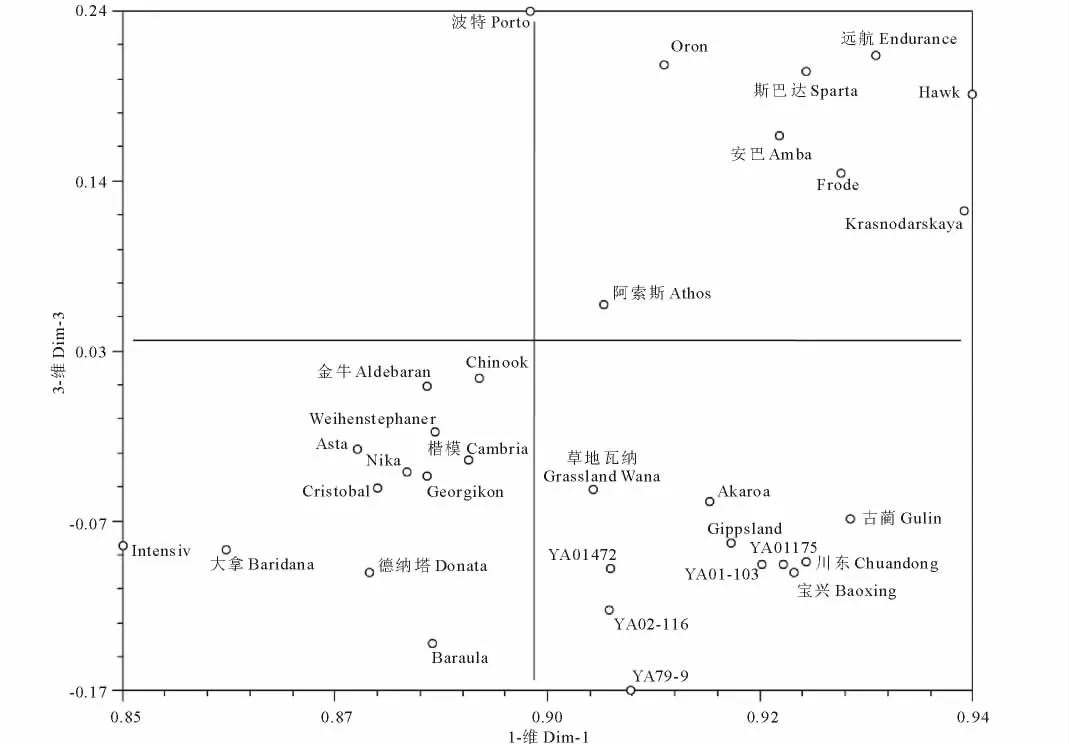
图3 基于SCoT谱型的32份鸭茅品种主成分分析Fig.3 Principal component analysis based on SCoT patterns in 32 orchardgrass varieties
我国是鸭茅的主要起源地之一,具有包括二倍体、四倍体和六倍体的野生鸭茅分布地26个[18-19],大量的研究表明我国野生鸭茅种质资源遗传多样性丰富。谢文刚等[23]对西南区鸭茅种质研究也表明我国鸭茅野生种质资源丰富,保持了较大的变异度和遗传多样性;万刚等[25]对鸭茅栽培驯化品种与野生材料遗传多样性的SSR研究分析表明,我国目前推广应用的鸭茅栽培驯化品种少且遗传基础狭窄,而我国的野生鸭茅种质资源具有丰富的遗传多样性,可通过开展相关的杂交筛选等工作,选育出能够满足我国不同生态条件下的鸭茅品种。Peng等[24]和钟声[20]的研究同样也证明了上述结论。本研究表明,鸭茅引进品种基因多样性指数等指标都高于栽培驯化品种。同时,目前国外已登记鸭茅品种达500个,在今后我国的鸭茅新品种选育策略上,应更多的针对于不同倍性、抗病、抗旱、叶量、适应性等特性进行杂交亲本材料选择,特别是在鸭茅易感的锈病等症状方面,选出在不同特性上表现突出和较弱的材料进行杂交,通过采用较为合理的育种方法,选育出适合我国不同生境生长的鸭茅新品种。另外,鸭茅为高度异花授粉植物[14],天然的杂交会引起品种内部的分化,对于品种的有效隔离也是一个必不可少的保护措施。
[1]李杰勤,王丽华,詹秋文,等.20个黑麦草品系的SRAP遗传多样性分析[J].草业学报,2013,22(2):158-164.
[2]韩国辉,向素琼,汪卫星,等.柑橘SCoT分子标记技术体系的建立及其在遗传分析中的应用[J].园艺学报,2011,38(7):1243-1250.
[3]Collard BCY,Mackill D J.Start codon targeted(SCoT)polymorphism:A simple,novel DNAmarker technique for generating genetargeted markers in plants[J].Plant Molecular Biology Reporter,2009,27:86-93.
[4]何庆元,王吴斌,杨红燕,等.利用SCoT标记分析不同秋眠型苜蓿的遗传多样性[J].草业学报,2012,21(2):133-140.
[5]Luo C,He X H,Chen H,et al.Analysis of diversity and relationships amongmango cultivars using start codon targeted(SCoT)markers[J].Biochemical Systematics and Ecology,2010,38:1176-1184.
[6]曾汉元,魏麟,刘鹏,等.能源草芦竹遗传多样性的ISSR分析[J].草业学报,2013,22(3):266-273.
[7]曾亮,袁庆华,王方,等.冰草属植物种质资源遗传多样性的ISSR分析[J].草业学报,2013,22(1):260-267.
[8]熊发前,蒋菁,钟瑞春,等.目标起始密码子多态性(SCoT)分子标记技术在花生属中的应用[J].作物学报,2010,36(12):2055-2061.
[9]Guo D L,Zhang JY,Liu CH.Genetic diversity in some grape varieties revealed by SCoT analyses[J].Molecular Biology Reports,2012,39:5307-5313.
[10]Luo C,He X H,Chen H,etal.Genetic relationship and diversity of Mangifera indica L.:revealed through SCoT analysis[J].Genetic Resources and Crop Evolution,2012,59:1505-1515.
[11]Xiong FQ,Zhong R C,Han ZQ,etal.Start codon targeted polymorphism for evaluation of functional genetic variation and relationships in cultivated peanut(Arachis hypogaea L.)genotypes[J].Molecular Biology Reports,2011,38:3487-3494.
[12]Gorji A M,Poczai P,Polgar Z,etal.Efficiency of arbitrarily amplified dominantmarkers(SCOT,ISSR and RAPD)for diagnostic fingerprinting in tetraploid potato[J].American Journal of Potato Research,2011,88:226-237.
[13]Lindner R,Garcia A.Geographic distribution and genetic resources of Dactylis in Galicia(northwest Spain)[J].Genetic Resources and Crop Evolution,1997,44:499-507.
[14]Bushman B S,Larson SR,Tuna M,et al.Orchardgrass(Dactylis glomerata L.)EST and SSR marker development,annotation,and transferability[J].Theoretical and Applied Genetics,2011,123:119-129.
[15]Lumaret R.Cytology,genetics,and evolution in the genus Dactylis[J].Critical Reviews in Plant Sciences,1988,7:55-89.
[16]Sanada Y,Tamura K,Yamada T.Relationship between water-soluble carbohydrates in falland spring and vigor of spring regrowth in orchardgrass[J].Crop Science,2010,50:380-390.
[17]Jafari A,Naseri H.Genetic variation and correlation among yield and quality traits in cocksfoot(Dactylis glomerata L.)[J].Journal of Agricultural Science,2007,145:599-610.
[18]彭燕,张新全.鸭茅种质资源多样性研究进展[J].植物遗传资源学报,2003,4(2):179-183.
[19]Stebbins G L,Zohary D.Cytogenetics and Evolutionary Studies in the Genus Dactylis L.Morphology,Distribution and Interrelationships of the Diploid Subspecies[M].California:University of California Press,1959,31:1-40.
[20]钟声.野生鸭茅杂交后代农艺性状的初步研究[J].草业学报,2007,16(1):69-74.
[21]张新全,杜逸,郑德诚.鸭茅二倍体和四倍体PMC减数分裂,花粉育性及结实性的研究[J].中国草地,1996,(6):38-40.
[22]帅素容,张新全,白史且.不同倍性鸭茅同工酶比较研究[J].草业科学,1998,(6):11-16.
[23]谢文刚,张新全,马啸,等.中国西南区鸭茅种质遗传变异的SSR分析[J].草业学报,2009,18(4):138-146.
[24]Peng Y,Zhang X Q,Deng Y L,et al.Evaluation of genetic diversity in wild orchardgrass(Dactylisglomerata L.)based on AFLP markers[J].Hereditas,2008,145:174-181.
[25]万刚,张新全,刘伟,等.鸭茅栽培驯化品种与野生材料遗传多样性比较的SSR分析[J].草业学报,2010,19(6):187-196.
[26]Doyle J J,Doyle JL,Brown A H D.Analysis of a polyploid complex in glycine with chloroplast and nuclear DNA[J].Australian Systematic Botany,1990,3:125-136.
[27]Yeh F C,Yang R C,Boyle T.POPGENE VERSION 1.31.Microsoft Windows-based Freeware for Population Genetic Analysis.Quick User Guide[M].University of Alberta:Center for International Forestry Research,1999.
[28]Nei M.Analysis of gene diversity in subdivided populations[J].Proceedings of the National Academy of Sciences of the United States of America,1973,70:3321-3323.
[29]Excoffier L,Smouse PE,Quattro JM.Analysis ofmolecular variance inferred from metric distances among DNA haplotypes:application to human mitochondrial DNA restriction data[J].Genetics,1992,131(2):479-491.
[30]张富民,葛颂.群体遗传学研究中的数据处理方法I.RAPD数据的AMOVA分析[J].生物多样性,2002,10:438-444.
[31]Pavlicek A,Hrda S,Flegr J.FreeTree-free ware program for construction of phylogenetic trees on the basis of distance data and bootstrap/jackknife analysis of the tree robustness.Application in the RAPD analysis of the genus Frenkelia[J].Folia Biologica,1999,45(3):97.
[32]NeiM,LiW H.Mathematicalmodel for studying the genetic variation in terms of restriction endonucleases[J].Proceedings of the National Academy of Sciences of the United States of America,1979,76:5269-5273.
[33]Huson D H,Scornavacca C.Dendroscope 3:An interactive viewer for rooted phylogenetic trees and networks[J].Systematic Biology,2012,61:1061-1067.
[34]Rohlf F J.NTSYS-pc:Numerical Taxonomy and Multivariate Analysis System,version 2.1.User Guide[M].New York:Exeter Software,2000.
[35]Luo C,He X H,Chen H,etal.Genetic diversity ofmango cultivars estimated using SCoT and ISSRmarkers[J].Biochemical Systematics and Ecology,2011,39:676-684.
