系统性红斑狼疮分子病因研究进展①
2013-11-27兰州大学第二医院肾内科兰州730030
曹 平 赵 玉 王 静 (兰州大学第二医院肾内科,兰州730030)
系统性红斑狼疮(Systemic lupus erythematosus,SLE)是一种多系统损伤的自身免疫性疾病,以T、B细胞异常活化、补体激活、免疫复合物清除障碍、沉积、自身抗体的产生及免疫调节异常为特点[1,2]。世界流行病学调查为1.4% ~21.9%,发病率为(7.4~159.4)/10万人,可造成多系统的损害,其中狼疮肾炎(Lupus nephritis,LN)是最严重的并发症之一,累积发病率亚洲人群最高,约为55%,非洲人群51%,其他各洲相对低,这些病人中23%约10年后发展为终末期肾病[3]。目前SLE病因、发病机理十分复杂,而且不同个体其发病机理不全相同[4]。近年越来越多的研究表明,机体的遗传因素在SLE的易感性发病方面处于主导地位,且是多基因协调控制的,其发生、发展与否有赖于环境因素的“扳机”及促进作用[5]。目前已知多种遗传基因与人类SLE易感性有关联,其中FEN1基因的单核苷酸多态性与SLE有关[5],FEN1基因异常可造成细胞凋亡机制障碍,诱发自身抗体形成最终导致疾病的发生发展[6]。另外,全基因组扫描证实,在人类染色体上,存在着50多个与SLE相关的区段,在SLE家族中呈现不同程度的连锁,提示其上可能存在着免疫相关的候选基因,包括人类白细胞抗原(HLA)Ⅰ类基因区 HLA-A、B、C、E、F、G、H、J和 X;Ⅱ类基因包括 HLA-DR、HLA-DQ、HLA·DP、TAP、LMP、DM;Ⅲ类基因包括编码补体成分基因(C2、C4A、C4B和Factor B)、细胞因子基因[肿瘤坏死因子(TNF)、α、β基因]、淋巴毒素(LTA、LTB)、热休克蛋白HSP70、T 细胞受体 (TCR)基因、TRIM、IL-4R、EGR1、KLRC1基因,人群16q12区(58.46 cM)OAZ基因和凋亡相关基因等[3,7-12],现就近几年报道SLE的可能的分子病因及发病机理作一概述。
1 FcγRII和 FcγⅢR(1q23) 与 SLE
FcγR家族属于免疫球蛋白超家族,其中FcγRIIa、FcγIIRb、FcγRⅢa 和 FcγRⅢb 存在基因多态性,被认为可能与SLE发病过程中免疫复合物清除有关,影响炎症反应,导致个体对自身免疫性疾病有不同易感性,该基因多态性与SLE发病的关系在不同的种群中研究结果不一致,Karassa等通过Meta分析得出FcγRIIa-R131是SLE发病的重要危险因素,但不是狼疮性肾炎的易感基因,且不同种族间没有差异性分布;FcγRⅢa-158F与LN有关,但与SLE无关[13]。Chu等对日本、泰国、中国人群也作了FcγR基因多态性SLE发病关系的Meta分析却得出FcγRIIa-R131与SLE和LN都无关的结论,而FcγRⅢa-158F与SLE有关,与LN发病无关[14]。这些研究提示FcγR基因确实是某些种群SLE和LN的易感基因,且种群间存在差别。为了更进一步的研究FcγR基因多态性在SLE发病中的作用,应进行多种人群采取更大规模的SLE遗传学和FcγR各亚型功能的研究。
2 补体缺陷与SLE
在SLE病人中,约50% ~75%病人存在低补体血症,很多患者的肾组织及皮肤组织亦可见大量的补体C1、C3和C4沉积[15]。研究发现,补体基因遗传性缺陷可致的低补体血症可能与SLE发病有关,尤其是经典途径中早期补体成份(C1~C4)缺失或无功能时容易出现狼疮样症状,补体基因遗传性缺陷可能导致补体缺乏溶血活性、膜攻击复合物(MAC)缺陷、免疫复合物清除障碍、凋亡细胞清除障碍[15]。肝脏合成补体能力下降、严重的肾脏病造成补体丢失等因素也可与SLE补体异常有关。单补体成分C3、C4及总补体(CH50)活性在疾病活动期均可降低,其中任何一种补体成分缺陷均可使CH50降低,补体水平下降作为SLE活动性指标之一[16,17]。临床上发现部分C3缺陷患者伴有膜增殖性肾小球肾炎、血尿或蛋白尿等表现,认为C3缺陷与一种称为C3肾炎因子的物质有关,现已确定C3肾炎因子为一种抗C3bBb复合物上新抗原的特异性IgG抗体,它起到稳定C3bBb活性的作用[17]。在临床上CH50测定是最简单的补体活性筛选方法。Lyon等报道了1例C2缺陷年轻患者:初诊表现为面部皮疹和颈部淋巴结肿大而无狼疮和其他自身免疫病的证据,2年后检测到抗核抗体和抗Sm抗体,最终诊断为SLE[18]。Jonsson等研究也证实了C2缺陷与SLE相关[19]。血浆C4是由C4A和C4B基因编码的蛋白产物,C4A和C4B的功能不同,等位基因C4AQ0和SLE的相关证据强于C4BQ0与SLE,目前大量研究表明C4AQ0与SLE人群显著相关,此等位基因大部分存在于DR3-B8单倍型[20]。总之,补体缺陷与SLE发病及疾病活动性关系密切,可在一定程度上反映病情的变化,CH50、C3和C4水平及其变化可以作为判断SLE疾病活动性的重要指标,可为SLE患者生物靶向治疗提供线索。
3 FEN1与SLE
FEN1基因位于人类染色体11q12,长1 144 bp,含有一个外显子,编码380个氨基酸,构成FEN1核酸酶的一级结构,是一种结构特异性多功能核酸酶[21],在DNA的多条代谢途径、维护染色体的稳定及细胞凋亡过程中发挥着重要作用[22,23]。Li Zheng等建立FEN1基因E160D点突变小鼠模型,造成FEN1约90%以上的 EXO和GEN酶活性丧失,在FEN1基因E160D点突变小鼠肾脏及肺组织中,TUNEL-阳性的核酸产物计数较野生型小鼠显著增高,表明FEN1基因E160D点突变可导致小鼠凋亡DNA产物增多。进一步分析发现,野生型小鼠成纤维细胞50%的凋亡细胞被转运所需要的时间比FEN1基因E160D突变小鼠50%的凋亡细胞转运时间少约24小时,后者对凋亡DNA产物的清除延迟,从而推测FEN1基因E160D点突变影响FEN1核酸酶功能,致使细胞凋亡过程中核小体DNA链不能有效分解,当大量的凋亡小体在机体组织中滞留时,作为具有免疫原性的靶抗原,构成危险信号对免疫系统造成威胁[24]。冯玲等对43例LN患者和26例健康者的外周血标本,采用全血基因组DNA柱式试剂盒提取DNA,直接PCR方法扩增FEN1基因片段,扩增后PCR产物应用基因测序方法检测FEN1基因序列,并对测序结果与基因数据库中FEN1基因进行比较,搜索可能的突变位点,结果发现LN患者正向测序中存在946位碱基C缺失突变(P=0.046),为该基因可能在LN的发病机制中的作用提供了一定的线索[25]。而我们初步研究中显示该基因外显子位点61563302、61563303碱基GT变为TC。我们认为FEN1在细胞周期或DNA损伤修复过程中会发生不同的翻译后修饰,这些翻译后修饰会抑制或促进它与不同蛋白之间的相互作用或者触发其降解。因此,我们推测FEN1在DNA复制或修复过程中所起的作用是受一系列的翻译后修饰调控的,翻译后修饰的改变会使FEN1功能失常,引起DNA复制和修复缺陷并最终导致疾病的发生,为此下一步我们继续应运基因操作技术就SLE患者该基因的多态性、表达产物等与疾病发生发展做更一步探讨。
4 细胞凋亡缺陷与SLE
细胞凋亡是一种相关基因严格调控的细胞生理性死亡,它是许多生物进程的一个重要组成部分。一般认为,有序的细胞凋亡能阻止凋亡细胞的继发性坏死和酶的释放,从而抵制炎症和免疫应答的发生,如果凋亡细胞清除障碍不能被及时清除,导致凋亡物质“进出”平衡失调,造成凋亡物质的累积,使细胞膜的完整性受损,细胞膜破裂,释放出大量修饰过的细胞核和细胞质物质,成为维持自身免疫应答的反复性的刺激原,不断攻击已经致敏的免疫系统,破坏其对自身抗原的中枢和外周耐受,出现组织器官受损(图1),进而引发炎症和免疫应答,促使SLE的发生、发展[26]。SLE患者细胞凋亡异常及凋亡产物清除障碍导致核小体过度释放,核小体与抗核小体抗体(AnuA)形成的免疫复合物沉积于肾小球上,AnuA在肾小球基底膜沉积是以固有细胞凋亡产生的核小体作为肾小球基底膜内的原位抗原或循环中核小体先与GBM相结合作为种植抗原,然后再在原位结合AnuA形成免疫复合物,这种沉积主要发生在上皮下,与膜性肾小球肾炎的发生发展有关,AnuA可能还参与Ⅰ型胶原的形成,从而加速肾小球的纤维化进程,另外LN患者体内免疫相关细胞,如淋巴细胞凋亡的增加,出现免疫系统紊乱,造成核小体过度释放,血循环中出现的DNA以寡核小体形式存在,抗核小体抗体与抗DNA抗体有交叉反应,这两者在LN的发生发展中都有着重要的作用[3,27,28]。因此,目前 AnuA 作为SLE患者肾脏损害的一项早期监测指标,将有助于早期干预治疗,提高预后。目前已知促进凋亡的基因有Fas及Fas配体(Fas-L)基因、p53基因、c-myc基因等;抑制凋亡的基因有bc1-2基因、bc1-xL基因、Pim-1基因等,促进基因与抑制基因的相互平衡作用决定着细胞凋亡与否,其中起关键作用的是Fas基因、Fas-L基因及bc1-2基因。
Fas/Fas-L基因突变可导致细胞凋亡活化途径受到抑制,使正常的免疫下调失效,自身反应性B细胞在体内积累增多,并产生致病性抗体,在SLE发病和病情进展中的重要作用已得到公认。Fas基因突变动物模型lpr/lpr小鼠在6月龄时发生SLE,而Fas-L基因突变gld/gld小鼠,也可以发生类似的疾病[29]。
bc1-2基因是细胞凋亡的抑制基因,其表达产物bc1-2蛋白是一种抑制多种细胞凋亡的蛋白,目前已有足够的证据说明在SLE外周T细胞中存在bc1-2表达的增多,且在各亚群间无明显差异,相反,B细胞则无此特性。与Fas基因相似,bc1-2基因异常也促进SLE的发生。首先,显著增多的bc1-2蛋白一方面可使T细胞逃避细胞凋亡;另一方面它又促进T细胞增殖,导致反应性T细胞的大量产生及增殖。其次,bc1-2基因的突变也导致其表达的异常。动物实验表明来自bc1-2基因突变鼠的T细胞对多种杀伤因素具有耐受性,这同样可过度抑制T细胞的正常凋亡。另外bc1-2基因与c-myc基因的联合表达也导致自反应性T细胞的出现及T细胞的过度增生[29]。最近我们收集了我省多家医院的LN患者肾脏活检组织标本40余例,对其中11例细胞凋亡检测发现存在凋亡小体堆积,见图2,其余标本正在实验过程中。
总之,细胞凋亡研究是一个方兴未艾的研究领域,是现代生物学的支柱学科之一。这方面的研究对包括SLE在内的多种疾病的发病机制提供了新的认识角度,为多种疾病的治疗提供了新思路和新途径。
5 微小干扰RNA与SLE
近年来,微小干扰(microRNA)作为一种非编码RNA分子,已有证据表明其在先天免疫应答及炎症因子的信号传导过程中起着重要的负调节作用,SLE干扰素通路异常活化可能与某些miRNA异常表达有关,并且在SLE重要的致病通路中起重要作用,Toll样受体7受到刺激后诱发浆细胞样树突状细胞(plasmactoid dendritic cell pDC)内19miRNAs产生不同程度的表达,其中miR-155和miR-155*主要的产物通过相反作用调节pDC介导Ⅰ型干扰素的产生,miR-155*通过靶向IRAKM,促进Ⅰ型干扰素的产生,而miR-155通过靶向TAB2,抑制Ⅰ型干扰素的产生,且在不同阶段发挥作用,此外,通过对miR-155和 miR-155*产生机制的研究,发现pDC自身分泌的Ⅰ型干扰素以及被激活的KHSRP蛋白可以在转录后水平反向调控miR-155和miR-155*的产生,这一结果解释了来自于同一前体的miR-155*和miR-155却能在不同的时间点被诱导的原因[30]。罗晓兵等研究小组采用候选基因测序的策略,选择大样本的病例-对照研究方法,在多个人群中发现miR-146a启动子区域存在基因变异(rs57095329)与SLE显著相关,携带疾病相关等位基因的个体,其miR-146a基因表达水平显著低于对照组,miR-146a表达缺陷,影响SLE易感基因ETS1编码表达的蛋白转录因子的结合能力,增加或者降低ETS1表达水平,导致miR-146a启动子活性的改变,也证实ETS1与miR-146a相互间的影响作用,在SLE发病中发挥叠加效应,引起Ⅰ型干扰素通路过度活化,进而参与SLE疾病的发生发展[31]。这些研究例证了复杂性疾病中多个易感基因通过相互作用共同参与SLE疾病的发生,阐明了新型有效的调控Ⅰ型干扰素产生环节的措施,可能是SLE治疗的潜在靶点。
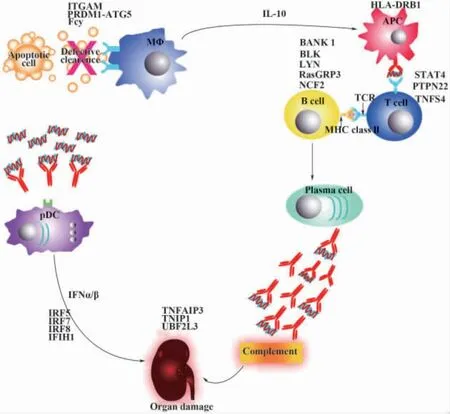
图1 系统性红斑狼疮免疫系统损害Fig.1 The impaired immune system in patients with systemic lupus erythematosus(SLE)
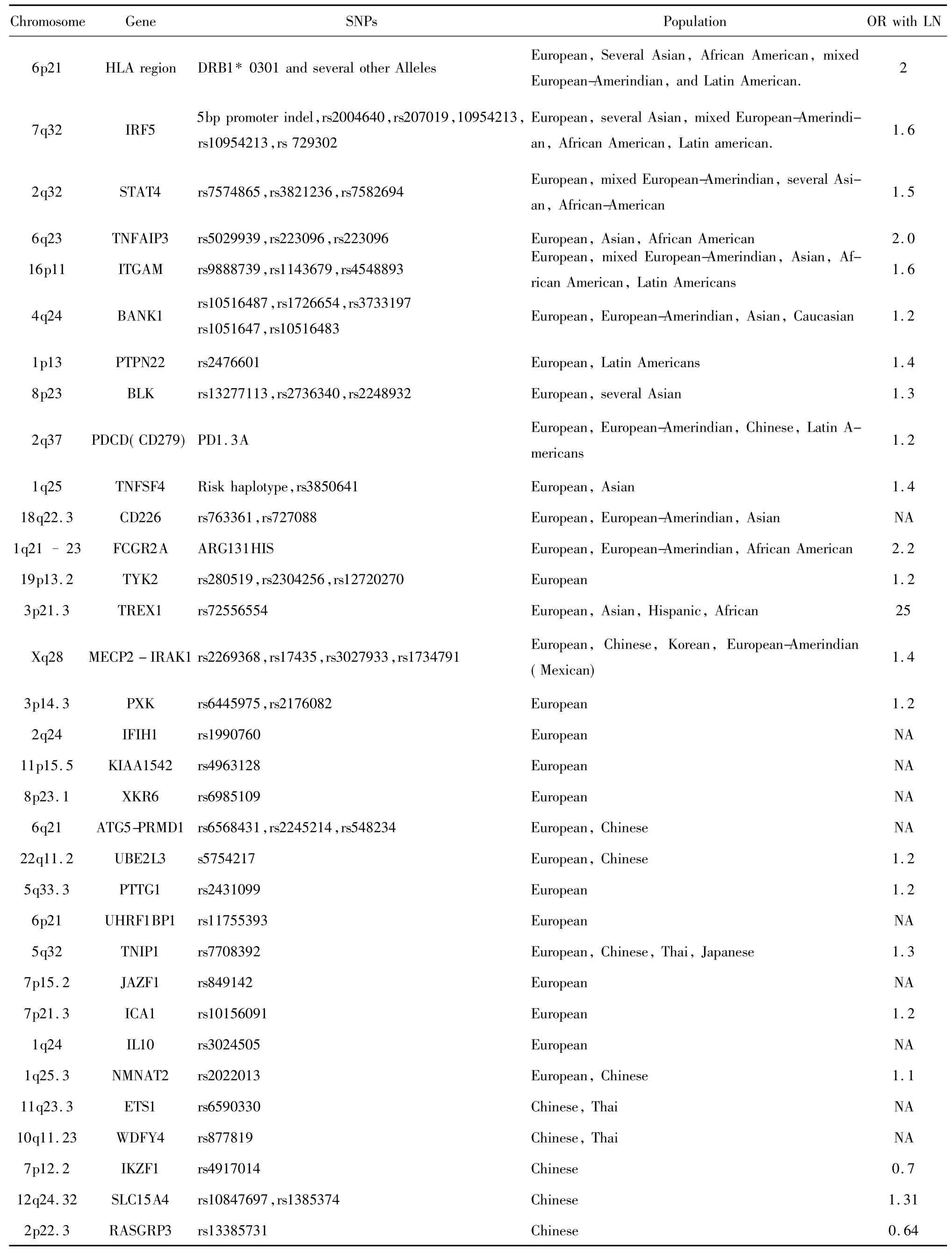
表1 SLE并发LN的易感基因Tab.1 Susceptibility genes in SLE associated with LN

图2 LN患者肾脏组织细胞凋亡检测图Fig.2 LN kidney tissue apoptosis detection figure
6 狼疮性肾炎易感基因
SLE基因学研究的发展促进了LN遗传和易感基因的研究的持续发展,LN病因、发病机理十分复杂,而且不同个体其发病机理不全相同,和SLE一致亦存在种族差异性和遗传异质性。以下列出近年来国内外研究已报道的不同基因多态性和LN的相关性[3],详见表 1。
7 展望
随着人类基因组计划的完成,医学基因组学研究以及计算机技术的快速发展,以当前的相关技术研究单基因遗传病的致病基因已不再是难题。但单基因遗传病的发病率通常比较低,严重危害人类健康和生活质量并困扰着科学研究策略的往往是一些多基因遗传病,如SLE,对其易感基因的研究不仅对于发现易感人群、进行基因诊断有重要价值,而且对于探索疾病的发病机制和开辟新的治疗途径也有重大的意义。
目前SLE分子病因研究的困难在于:(1)多因素复杂性,十多个或者数十个甚至更多基因在不同环境下存在叠加交互作用。(2)遗传异质性研究的疾病可能是存在不同遗传背景表型集合。(3)所选择的候选基因假阳性不一定和SLE疾病真实相关。(4)临床疾病的诊断和分类的不确定性,因表型而被错误地作为患病的研究对象及未经随访证实的健康对照。(5)由于研究人群的遗传结构不一致、样本量不同、多重检验而导致的假阳性的结果会造成很多误导。
近年来,有关SLE发病病因和诊断的研究虽然进展较大,但仍缺乏多中心、多对照的研究方法,各地科研侧重点有所不同,难以从总体上把握其发病病因,尚有待于充分借鉴现代科学技术,加大基础研究力度,力求从细胞、分子水平阐明SLE的病因,为临床治疗提供可靠的理论依据。糖皮质激素和免疫抑制剂联合治疗已经使SLE患者的预后及社会回归率得到了很大的提高,但SLE仍然缓解率低下,部分重症患者对激素及免疫抑制剂治疗无效或疗效欠佳,长期使用带来的副作用也严重威胁了这些患者的健康,提高SLE的疗效,改善患者的预后迫切需要我们对SLE发病机制的进行深入的理解,诸如抗CD20单抗(rituximab,利妥昔单抗)、抗CD5、抗IL-6、抗IL-10、干扰素等抗体,这些针对SLE发病机制中某一环节或影响其疾病进展的关键分子进行的选择性靶向治疗,俨然已经成为治疗SLE的新方向,因此,继续针对在SLE发病中起关键作用的遗传流行病学、基因易感区域定位、克隆、转录、翻译后的修饰等环节的深入研究,是最终提高SLE疗效、控制病变进展、改善预后的根本途径,基于此以生物技术为基础的多种生物制剂的研发和应用将是控制SLE的有效措施。
1 Foster M H.T cells and B cells in lupus nephritis[J].In:Semin Nephrol,2007;27:47-58.
2 Goodnow C C,Sprent J,de St Groth B F et al.Cellular and genetic mechanisms of self tolerance and autoimmunity[J].Nature,2005;435(7042):590-597.
3 Alberto de Zubiria Salgado,Catalina Herrera-Diaz.Lupus nephritis:an overview of recent findings[J].Autoimmune Dis,2012;2012:849684.
4 Crispin J C,Liossis S N,Kis-Toth K et al.Pathogenesis of human systemic lupus erythematosus:recent advances[J].Trends Mol Med,2010;16(2):47-57.
5 Kim I,Hur Nw Fau-Shin H D,Park B L et al.Associations of DNase IV polymorphisms with autoantibodies in patients with[J].Rheumatology(Oxford),2008;47(7):996-999.
6 Zheng L,Dai H F,Zhou M et al.Fen1 mutations result in autoimmunity,chronic inflammation and cancers[J].Nat Med,2007;13(7):812-819.
7 Corporaal S,Bijl M F,Kallenberg C G.Familial occurrence of autoimmune diseases and autoantibodies in a Caucasian[J].Clin Rheumatol,2002;21(2):108-113.
8 Wong M,Tsao B P.Current topics in human SLE genetics[J].Springer Semin Immunopathol,2006;28(2):97-107.
9 Harley J B,Kelly J A,Kaufman K M.Unraveling the genetics of systemic lupus erythematosus[J].Springer Semin Immunopathol,2006;28(2):119-130.
10 Morel L.Genetics of human lupus nephritis[J].Semin Nephrol,2007;27(1):2-11.
11 Rizzo R,Hviid Tv Govoni M,Padovan M et al.HLA-G genotype and HLA-G expression in systemic lupus erythematosus:HLA-G as a putative susceptibility gene in systemic lupus erthematosus[J].Tissue Antigens,2008;71(6):520-529.
12 Ramos P S,Brown E E,Kimberly R P et al.Genetic factors predisposing to systemic lupus erythematosus and lupus nephritis[J].Semin Nephrol,2010;30(2):164-176.
13 Brambila-Tapia Aj,Davalos-Rodriguez IP.Fcgamma receptor polymorphisms and systemic lupus erythematosus[J].Rev Invest Clin,2009;61(1):66-72.
14 Niederer H A,Clatworthy M R,Willcocks L C et al.FcgammaRIIB,FcgammaRIIIB,and systemic lupus erythematosus[J].Ann N Y Acad Sci,2010;1183:69-88.
15 Pettigrew H D,Teuber S S,Gershwin M E.Clinical significance of complement deficiencies[J].Ann N Y Acad Sci,2009;1173:108-123.
16 Sarma J V.Ward P A.The complement system.Cell Tissue Res,2011;343(1):227-235.
17 Wen L,Atkinson J P,Giclas P C.Clinical and laboratory evaluation of complement deficiency[J].J Allergy Clin Immunol,2004;113(4):585-593;quiz 594.
18 Lyon V B,Nocton J J,Drolet B A et al.Esterly NB:Necrotic facial papules in an adolescent:C2 deficiency with eventual development[J].Pediatr Dermatol,2003;20(4):318-322.
19 Jonsson G,Truedsson L,Sturfelt G et al.Hereditary C2 deficiency in Sweden:frequent occurrence of invasive infection[J].Medicine(Baltimore),2005;84(1):23-34.
20 Graham R,Ortmann W,Rodine P et al.Specific combinations of HLA-DR2 and DR3 class II haplotypes contribute graded risk for disease susceptibility and autoantibodies in human SLE[J].European J Human Genetics,2007;15(8):8.
21 Ayyagari R,Gomes X V,Gordenin D A et al.Okazaki fragment maturation in yeast.I.Distribution of functions between FEN1[J].J Biol Chem,2003;278(3):1618-1625.
22 Liu Y,Kao H I,Bambara R A.Flap endonuclease 1:a central component of DNA metabolism[J].Annu Rev Biochem,2004;73(7):589-615.
23 Parrish J Z,Yang C,Shen B et al.CRN-1,a caenorhabditis elegans FEN-1 homologue,cooperates with CPS-6/Endo.G to promote apoptotic DNA degradation [J].Embo J,2003;22(13):3451-3460.
24 Zheng L,Zhou M,Chai Q et al.Novel function of the flap endonuclease 1 complex in processing stalled DNA[J].EMBO Rep,2005;6(1):83-89.
25 冯 玲,王 静,王俭勤.FEN1基因突变在狼疮性肾炎发病机制中的作用[J].国际泌尿系统杂志,2010;30(5):625-628.
26 Guerra S G,Vyse T J,Cunninghame Graham D S.The genetics of lupus:a functional perspective[J].Arthritis Res Ther,2012;14(3):211.
27 Kalaaji M,Fenton K A,Mortensen E S et al.Glomerular apoptotic nucleosomes are central target structures for nephritogenic[J].Kidney Int,2007;71(7):664-672.
28 王 静,王志平.FEN1基因突变在狼疮肾炎凋亡核小体清除机制中的作用[J].中华肾脏病杂志,2011;27(7):538-540.
29 Paunovic V,Carter N A,Thalhamer T et al.Immune complex-mediated co-ligation of the BCR with FcgammaRIIB results in[J].J Autoimmun,2012;39(4):332-346.
30 Zhou H,Huang X,Cui H et al.miR-155 and its star-form partner miR-155* cooperatively regulate type I[J].Blood,2010;116(26):5885-5894.
31 Luo X,Yang W,Ye D Q et al.A functional variant in microRNA-146a promoter modulates its expression and[J].PLoS Genet,2011;7(6):e1002128.
