漆酶/天然介体体系漂白硫酸盐竹浆的机理研究
2012-12-31周生飞詹怀宇姚向荣付时雨
周生飞 詹怀宇 姚向荣 付时雨
(华南理工大学制浆造纸工程国家重点实验室,广东广州,510640)
漆酶/天然介体体系是一种对环境友好的生物反应体系,在制浆造纸中有着重要的应用。在二次纤维回用中,它有助于废纸脱墨,提高脱墨浆的可漂性,同时提高纸浆的裂断长和撕裂指数;在高得率制浆中,它能显著降低能耗,提高成纸强度;在化学法制浆中,它能降低纸浆卡伯值,提高白度;在纸浆漂白中,它能取代传统的含氯漂白,降低有毒和致癌物质的排放量,减少对环境的污染和人类的危害;在造纸废水处理中,它能降解造纸废水中的有毒物质和有色物质。此外,它还能用于改善纸浆纤维性能,提高纸张湿强度等[1]。
笔者曾用羟基苯甲酸、香草酸、对羟基苯甲醛、丁香酸、香草醛、丁香醛、乙酰丁香酮等7种小分子酚醛物质及它们的混合物作为漆酶/介体体系的天然介体,用于硫酸盐法竹浆的漆酶/介体体系漂白,这些体系都能不同程度地促进竹浆最终白度的提高和木素的脱除。然而,漆酶/天然介体体系的漂白机理至今仍不十分明确,很有必要继续深入探讨。本实验对硫酸盐竹浆进行了包含漆酶/天然介体体系漂白段的全无氯漂白,并对各段的残余木素进行了GPC、TGA、FT-IR、1H-NMR和13C-NMR分析,期望通过漆酶/天然介体体系漂白段前后竹浆残余木素的表征,探索漆酶/天然介体体系的漂白机理。
1 实验
1.1 原料和药品
硫酸盐竹浆,初始卡伯值16.0、白度30.3%。
主要药品和试剂:漆酶(通过实验室培养的白腐菌获得),混合纤维素酶(由CMC酶和Avicel酶组成,液体,夏盛公司提供),吡啶(分析纯),醋酸酐(分析纯),THF(色谱纯),KBr(光谱纯),无水乙醇(分析纯),DMSO-d6(含TMS)。
1.2 竹浆的TCF漂白
首先对竹浆进行了TCF漂白,漂序为O-LMS-QP,具体操作条件如下。
氧脱木素(O):在旋转式反应釜中进行,浆浓10%,NaOH用量4.0%,MgSO4用量0.5%,氧压0.5MPa,温度100℃,时间60min。
漆酶/天然介体体系处理(LMS):在PARR 4843高压反应釜中进行,所用天然介体为对羟基苯甲酸、香草酸、对羟基苯甲醛、丁香酸、香草醛、丁香醛、乙酰丁香酮等小分子酚醛物质的混合物,浆浓10%,漆酶用量5IU/g,介体用量2%,用醋酸-醋酸钠缓冲溶液调节pH值为4.0,温度50℃,氧压0.5MPa,反应时间120min,搅拌转速150r/min。
螯合处理(Q):将浆料及化学品密封于塑料袋内,揉搓均匀后置于恒温水浴锅中,浆浓10%,ED-TA用量1.0%,漂终pH值3.0,温度70℃,反应时间60min。
过氧化氢漂白(P):将浆料及化学品密封于塑料袋内,揉搓均匀后置于恒温水浴锅中,浆浓10%,H2O2用量3.0%,NaOH用量2.0%,MgSO4用量0.05%,EDTA用量0.2%,温度90℃,反应时间150min。
1.3 漂白各段竹浆残余木素的提取
首先用温和的酶水解-酸水解法[2]从竹浆中制备粗木素,然后用乙醚对粗木素进行提纯。
温和的酶水解:将湿润的竹浆置于三角瓶中,加入一定量的去离子水,浆浓约为3%;加入pH值4.8的醋酸-醋酸钠缓冲溶液,使浆中离子浓度为50mmol/L;加入混合纤维素酶,用量为50FPU/g(绝干浆),然后将三角瓶置于50℃的摇床内,在200r/min下振荡,24h后再次加入酶液,总反应时间为48h。反应结束后,将混浊液在4500r/min下离心10min,得到沉淀。
酸水解:将酶水解得到的沉淀加入到0.05mol/L HCl的二氧六环-水(前者与后者体积比85∶15,下同)中,在通N2保护的情况下微沸(103℃左右,油浴)回流2h;反应液冷却后用G2玻璃滤器过滤,残留固体用二氧六环-水洗涤至无色,合并滤液,在强烈搅拌下用NaHCO3中和;然后将液体缓慢滴入大量pH值为2的水中(水体积是液体体积10倍以上),出现淡黄色沉淀;在4500r/min下离心10min,得到沉淀,用一定体积的二氯甲烷洗涤3次后,冷冻干燥,制得粗木素。
粗木素提纯:将粗木素溶于二氧六环-水中,然后缓慢滴入大量乙醚(10倍体积以上)中,出现淡黄色沉淀;在4500r/min下离心10min,得到沉淀,用乙醚洗涤,冷冻干燥,得到纯化木素。
1.4 木素的乙酰化
将纯化木素置于带塞的三角瓶中,加入足量的吡啶和醋酸酐(前者与后者体积比1∶2),用N2排出三角瓶中的空气,置于暗中反应72h,反应期间不定期振荡。反应结束后,将液体缓慢滴入大量乙醚(10倍体积以上),析出浅黄色的沉淀,即为乙酰化后的木素。离心分离,并用乙醚洗涤乙酰化后的木素至无吡啶味,冷冻干燥备用。
1.5 对木素的分析
木素的分子质量分布在Agilent 1200 series上进行测定,2根凝胶色谱柱为Agilent PL gel 5μm 103Å和500Å,RID检测器,流量1.0mL/min柱温30℃,标线用聚苯乙烯作出,其质均相对分子质量为1490~53500。
木素的TGA分析在TGA Q 500上进行,升温速率为20℃/min。
木素的FT-IR分析在Bruker Tensor 27上进行,采用KBr压片法,扫描次数为16次。
木素的1H-NMR分析和13C-NMR分析是先将乙酰化的木素溶于DMSO-d6(含TMS)中,然后在Bruker AV400 NMR上进行。
2 结果与讨论
2.1 竹浆木素的GPC分析
漂白各段竹浆残余木素的GPC分析结果如表1所示。由表1可知,经O段处理后,残余木素的质均分子质量略有增加,而经LMS段处理后,残余木素的质均分子质量明显变小,说明残余木素在漂白过程中的总体变化趋势是质均分子质量变小。残余木素分子质量分布经O段处理后,多分散性变小,说明分子质量分布变窄;经LMS段处理后,残余木素的分子质量分布反而变得更为分散。

表1 漂白各段竹浆残余木素的相对分子质量
2.2 竹浆木素的TGA分析
O段和LMS段残余木素的热重(TG)曲线及微分(DTG)曲线见图1。
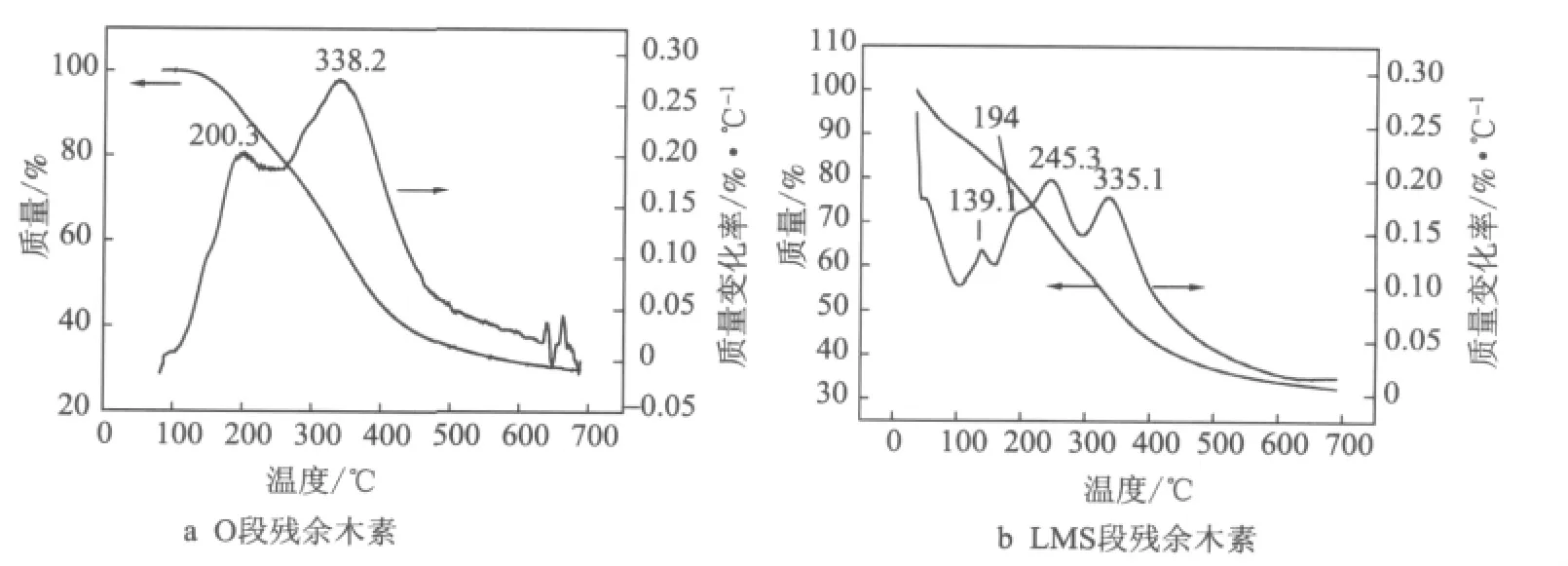
图1 O段/LMS段残余木素的TG和DTG曲线
由图1a可知,O段残余木素的热解过程分为3个区段:温度<210℃时,微分曲线在200.3℃处出现第一个失重峰,主要是样品中所含水分的去除;在210~600℃范围内,木素发生明显热解,微分曲线在338.2℃处有较大失重峰,这个峰区间为木素的主要热解阶段,木素失重达56.2%,600℃处木素的残余质量为31.6%;温度>600℃,热重曲线平缓,微分曲线在650℃附近出现小的失重峰,为木素的二次热解。
由图1b可知,LMS段残余木素的热解过程也分为3个区段:温度<159.5℃时,微分曲线在139.1℃处出现一个失重峰,主要是样品中所含水分的去除;159.5~600℃为木素热解的主要区间,木素发生明显热解,微分曲线在194℃处有一个失重峰肩,在245.3℃和335.1℃处有2个主要的失重峰;木素在159.5~298℃峰区间的失重为22.9%,在298~600℃峰区间的失重为25.8%,600℃处木素的残余质量为34.1%;温度>600℃,热重曲线平缓,微分曲线无失重峰。
综上所述,在LMS段处理前后,竹浆残余木素的热解行为明显不同。LMS段前残余木素仅在338.2℃处有一主要的失重峰,LMS段后残余木素除了在该处附近的335.1℃处有一个主要的失重峰外,还在较低温度245.3℃处出现一个更大的失重峰。可见,经LMS段后,竹浆残余木素的热稳定性变差,更易在较低温度下热解。
2.3 竹浆木素的FT-IR分析
图2为LMS段残余木素的FT-IR,各个吸收峰根据参考文献[3-9]确定,其归属见表2。

图2 LMS段残余木素的FT-IR

表2 竹浆残余木素FT-IR吸收峰的归属
3470cm-1说明原浆木素、O段和LMS段残余木素中均存在一定量的游离脂肪族羟基;3130cm-1说明原浆木素和LMS段残余木素中存在游离酚羟基,而O段残余木素中几乎没有游离酚羟基;3080cm-1说明O段残余木素中有少量—COOH。对于原浆木素和LMS段残余木素,3130cm-1比3470cm-1强,说明游离酚羟基比脂肪族羟基多。2940cm-1附近为CH3、CH2、CH的C—H伸缩振动,原浆木素和LMS段残余木素在此处的吸收类似,但是O段残余木素的CH3吸收显著减弱,说明O段中大量的—OCH3被脱除。1767cm-1为芳香乙酰基,1718cm-1为脂肪乙酰基,原浆木素在1767cm-1处有大的吸收峰,说明原浆木素中主要为酚羟基,脂肪族羟基少;O段残余木素在1743cm-1有大的吸收峰,处于1767~1718cm-1之间,说明乙酰化的酚羟基和脂肪族羟基的含量相当,结合3130cm-1处无游离酚羟基,说明O段残余木素脂肪族羟基比酚羟基稍多;LMS段残余木素在1743cm-1处有大的吸收峰且峰肩向1767cm-1延伸,说明LMS段残余木素以脂肪族羟基为主。综上所述,原浆木素羟基以酚羟基为主,O段残余木素中脂肪族羟基比酚羟基稍多,而LMS段残余木素则以脂肪族羟基为主,说明木素在O段和LMS段中,苯环被持续破坏,导致酚羟基减少,脂肪族羟基增多。
1685~1652cm-1为共轭CO的吸收,1685cm-1为共轭醛CO,1652cm-1为共轭酮CO;原浆木素中1685cm-1处和1652cm-1处的吸收相当,O段残余木素相对原浆木素在1652cm-1处的吸收增强,LMS段残余木素相对O段残余木素在1685cm-1处的吸收又增强,说明漂白过程中木素先生成共轭酮,然后又生成共轭醛。
对于原浆木素,在1465~1458cm-1、1418cm-1、1400cm-1处为愈创木基(G)上—OCH3的C—H弯曲振动,在1385cm-1、1372cm-1、1338~1331cm-1处为紫丁香基(S)上—OCH3的C—H弯曲振动;原浆木素中G单元—OCH3的吸收比S单元—OCH3的吸收略强,可得G与S的摩尔比大于2∶1,即S单元占木素结构单元的比例不到1/3(以物质的量计),竹子原浆木素的主要结构单元为G单元。
对于LMS段残余木素,在1465~1455cm-1、1419cm-1、1400cm-1处为G单元上—OCH3的C—H弯曲振动,在1385cm-1、1372cm-1、1333cm-1处为S单元上—OCH3的C—H弯曲振动;G单元的吸收比S单元稍弱,可得G与S的摩尔比小于2∶1,G单元仍为主要结构单元,但S单元占木素结构单元的比例相对原浆木素稍多,即经LMS段后,残余木素中S单元的含量增多。
在1045cm-1处为CC—O的C—O,在1013cm-1处为Ar-O的C—O,原浆木素和O段残余木素在该两处的吸收类似,LMS段残余木素在1045cm-1处吸收比原浆木素和O段残余竹浆木素强,而在1013cm-1处变弱,说明LMS段中,木素苯环被破坏,苯环转变成脂肪链。
2.4 竹浆木素的1H-NMR分析
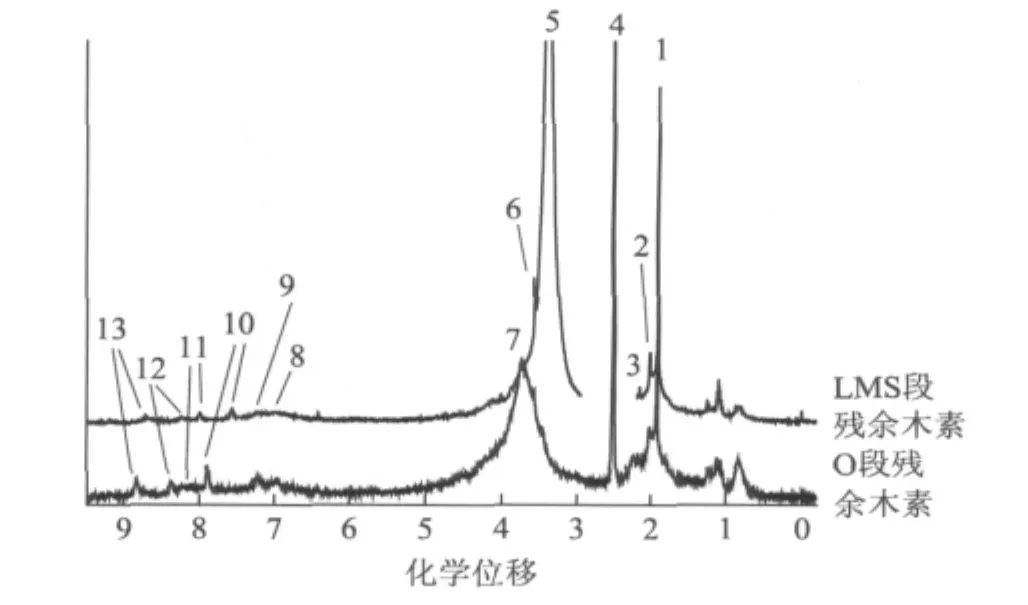
图3 O段/LMS段残余木素的1H-NMR图

表3 O段/LMS段残余木素1H-NMR谱共振峰的归属
O段和LMS段残余木素的1H-NMR图如图3所示,图3中各峰的归属根据参考文献[8-12]确定(见表3)。由表3可知,8#~13#峰为苯环质子的共振,5#~7#峰为—OCH3的质子共振。由8#~13#峰可知,这2种木素中都含有愈创木基(G)、紫丁香基(S)和对羟基苯基(H)这3种苯丙烷结构单元,LMS段前后竹浆残余木素为GSH型。1#峰和2#峰为脂肪乙酰基,3#峰为芳香乙酰基,O段残余木素中,1#峰和2#峰积分面积之和与3#峰积分面积之比为2.5∶1,LMS段残余木素中,它们的积分面积之比为3∶1,说明经LMS段后,脂肪乙酰基增多,即残余木素中脂肪族羟基增多,这是由于经LMS段后竹浆残余木素的破坏增多。5#峰为脂肪链—OCH3,6#峰和7#峰为苯环—OCH3,O段残余木素中以7#峰为主,而LMS段残余木素中以5#峰为主,说明LMS段残余木素中脂肪链—OCH3比O段残余木素的多、苯环—OCH3比O段残余木素的少,这是由于LMS段中木素破坏增加。
图4是O段和LMS段残余木素苯环质子区的比较,从O段到LMS段,代表G单元和S单元的峰的面积之比G/S和G’/S’均变大,说明经LMS段后,竹浆残余木素S单元相对含量增多;H/G和H’/G’之比均变小,说明经LMS段后,竹浆残余木素H单元相对含量减少。可见,漆酶/天然介体体系漂白对竹浆木素S单元的破坏较少,而对H单元的破坏和去除则较多。

图4 O段/LMS段竹浆残余木素1H-NMR的苯环质子区
对1H-NMR谱的相关区域进行积分,可以由积分面积计算出竹浆残余木素中主要基团的摩尔比,如表4所示。经LMS段后,残余木素每个苯丙烷单元对应的甲氧基数增多,这是因为在LMS段中部分苯环被破坏后残留在竹浆中,导致苯环数量减少,然而甲氧基却仍然保留;苯丙烷单元对应的脂肪族羟基显著增加,也说明LMS段中木素的苯环被进一步氧化破坏。

表4 木素中各基团的摩尔比
2.5 竹浆木素的13C-NMR分析
LMS段前后竹浆残余木素的13C-NMR谱如图5和图6所示,各峰的归属根据参考文献[4-5,9,11-15]确定(见表5)。
如图5所示,对于O段残余木素,1#峰为脂肪乙酰基的CO,2#峰为芳香乙酰基的CO,前者与后者的积分面积之比大于1,可见,在O段残余木素中,同时存在脂肪族羟基和酚羟基,脂肪族羟基的含量比酚羟基多。3#峰为酯化紫丁香基(S)的C-3和C-5的共振,4#峰为愈创木基(G)的C-3的共振,6#峰为愈创木基(G)的C-4的共振,7#峰为对羟基苯基(H)的C-4的共振,说明O段残余木素为GSH型,由3#峰、6#峰与7#峰的积分面积比为1∶2∶1可得,苯丙烷单元S、G、H的摩尔比为0.5∶2∶1(3#峰由2个C的共振产生,而6#峰和7#峰由1个C的共振产生),即O段残余木素以G单元为主,H单元含量多于S单元含量,说明在O段中—OCH3被大量脱除。
如图6所示,在LMS段残余木素中,1#峰与2#峰积分面积之比为1∶0.07,比值远远大于1,可见,同时存在脂肪族羟基和酚羟基,且以脂肪族羟基为主,酚羟基非常少。3#峰~7#峰、9#峰~11#峰为苯环C的共振,可知LMS段残余木素为GSH型。由化学位移为143.6(H)、148.0~149.7(G)、152.6(S)处的积分得,H、G、S的摩尔比为1∶4.94∶2.78。可见,LMS段残余木素以G为主;相对于O段残余木素,LMS段残余木素S单元增加,H单元减少,与前文FT-IR和13C-NMR的分析结果一致。


从图6的化学位移为75.3、63.2、62.0、60.7和59.3可知,木素单元之间的连接方式主要有β-1’,β-O-4’和β-5’,它们的积分面积比为1∶2.38∶0.26,则β-1’、β-O-4’和β-5’3种主要连接方式的摩尔分数(只考虑这3种连接时)依次为27.5%、65.4%、7.1%。在Sakakibara提出的由28个苯丙烷单元构成的针叶木木素的结构模型中,以β-O-4’连接为主,且有3个β-1’和4个β-5’连接;而Nimz提出的由25个苯丙烷单元构成的阔叶木木素模型中,β-1’连接为3个,β-5’连接仅有2个[16]。竹子作为禾本科植物,除以β-O-4’为主的连接外,还有较多的β-1’连接,而β-5’连接较少,这是合理的。

表5 LMS段前后竹浆残余木素13C-NMR谱中各峰归属
综上所述,竹浆残余木素中脂肪族羟基多于酚羟基,LMS段残余木素酚羟基很少。竹浆木素为GSH型,结构单元以G单元为主,O段残余木素中G>H>S(以物质的量计,下同),LMS段残余木素中G>S>H,说明经LMS段后,竹浆木素中S单元增多。苯丙烷单元之间的连接以β-O-4’为主,且有β-1’和β-5’连接,经LMS段后,竹浆木素中β-1’连接的含量增加,而β-5’连接很少。经LMS段后,竹浆残余木素的破坏加剧,脂肪族羟基增加,H单元的含量减少,G单元的含量略有减少,S单元的含量从27%增至31.9%。
3 结论
3.1 采用GPC分析了硫酸盐竹浆木素、O段和LMS段的残余木素。结果表明,原浆木素的质均相对分子质量为11010、O段残余木素的为12150、LMS段残余木素的为7374,多分散性依次为1.49、1.03和2.38,可见,随漂白(O、LMS)的进行,竹浆残余木素的总体变化趋势为分子质量降低。
3.2 采用TGA分析了O段和LMS段残余木素。结果表明,O段残余木素在338.2℃处有1个主要的失重峰,而LMS段残余木素在335.1℃处和245.3℃处有2个主要的失重峰。可见,经LMS段处理后,竹浆残余木素的热稳定性变差,更易在较低温度下热解。
3.3 采用FT-IR和1H-NMR分析了原浆木素、O段和LMS段残余木素。采用13C-NMR分析了O段和LMS段残余木素。结果表明,竹浆木素为GSH型,结构单元以G为主,苯丙烷单元之间的连接以β-O-4’为主,此外还有β-1’和β-5’连接。原浆木素中,羟基以酚羟基为主,苯丙烷单元中S的含量(以物质的量计,下同)少于1/3;O段残余木素中,脂肪族羟基比酚羟基多,并有少量—COOH、—OCH3脱除,苯丙烷单元中G>H>S;LMS段残余木素中,羟基以脂肪族羟基为主,S单元含量增加,H单元含量减少,β-1’连接增加,β-5’连接减少。综上所述,随漂白(O、LMS)的进行,—OCH3被脱除,苯环被破坏,脂肪族羟基增多;经LMS段后,竹浆残余木素中S单元增加,H单元减少。
[1]张爱萍,秦梦华,徐清华.漆酶在制浆造纸中的应用研究进展[J].中国造纸学报,2004,19(2):161.
[2]Argyropoulos D S,Sun Y,Palus E.Isolation of residual Kraft lignin in high yield and purity[J].J Pulp Pap Sci,2002,28(2):50.
[3]Lawther J M,Sun R C,Banks W B.Rapid isolation andstructural characterization of alkali-soluble ligninsduring alkaline treatment and atmospheric refining of wheat straw[J].Industrial Crops and Products,1996(5):97.
[4]Fernández-Bolaños J,Felizón B,Heredia A,et al.Characterization of the lignin obtained by alkaline delignificationand of the cellulose residue from steam-exploded olive stones[J].Bioresource Technology,1999,68:121.
[5]Sun R C,Tomkinson J,Wang S Q,et al.Characterization of lignins from wheat straw by alkalineperoxide treatment[J].Polymer Degradation and Stability,2000,67:101.
[6]付时雨.生物漂白过程木素结构的红外光谱分析[J].纤维素科学与技术,2000,8(2):30.
[7]泉美治,小川雅弥,加藤俊二,等.仪器分析导论:第4册[M].2版.李春鸿,刘振海,译.北京:化学工业出版社,2005:1.
[8]Ibrahim M N M,Norhidaya Z,Sipaut C S,et al.Chemical and thermal properties of lignins from oilpalm biomass as a substitute for phenol in a phenolformaldehyde resin production[J].Carbohydrate Polymers,2011,86(1):112.
[9]Sun X F,Jing Z,Fowler P,et al.Structural characterization and isolation of lignin and hemicelluloses from barley straw[J].Industrial Crops and Products,2011,33:588.
[10]García A,Toledano A,Serrano L,et al.Characterization of lignins obtained by selective precipitation[J].Separation and Purification Technology,2009,68:193.
[11]Oliveira L,Evtuguin D,Cordeiro N,et al.Structural characterization of stalk lignin from banana plant[J].Industrial Crops and Products,2009,29:86.
[12]She D,Xu F,Geng Z C,et al.Physicochemical characterization of extracted lignin from sweet sorghum stem[J].Industrial Crops and Products,2010,32:21.
[13]Sealey J,Ragauskas A J.Residual lignin studies oflaccase-delignifiedkraftpulps[J].Enzyme and Microbial Technology,1998,23:422.
[14]Ghatak H R.Spectroscopic comparison of lignin separated by electrolysisand acid precipitation of wheat straw soda black liquor[J].Industrial Crops and Products,2008,28:206.
[15]Hage R E,Brosse N,Chrusciel L.Characterization of milled wood lignin and ethanol organosolv ligninfrom miscanthus[J].Polymer Degradation and Stability,2009,94:1632.
[16]詹怀宇,李志强,蔡再生.纤维化学与物理[M].北京:科学出版社,2005:272.
