Mg-Li合金界面结构及合金化效应的第一性原理研究
2012-10-26韩培德白晋纲李洪飞张彩丽许并社
韩培德,白晋纲,李洪飞,张彩丽,许并社
(1.太原理工大学a.新材料界面科学与工程教育部重点实验室,b.材料科学与工程学院,太原 030024;2.太原钢铁(集团)有限公司 技术中心,太原 030003)
镁锂合金是最轻的金属结构材料,密度在1.35~1.65g/cm3,具有比强度和比刚度高、抗冷热变形能力强、低温性能好等特性。镁锂合金还具有抗高能粒子穿透能力强、电磁屏蔽性能好、阻尼性能好、切削加工性优良等特性,是航天、航空、电子和军事等领域理想的轻质结构材料。但镁锂合金也存在绝对强度低、耐高温能力差、抗蠕变性能不足和抗腐蚀性差等缺点。欧美一些国家对镁锂合金的研究起步较早,并在航空航天等领域获得了广泛应用。我国对镁锂合金的研究近年来取得了很大进展,但开发应用尚处于初级阶段。
镁锂二元合金中Li的质量分数小于5.5%时只有α相,大于10.9%时只有β相,中间为α、β两相共存区[1]。合金化是提高金属材料性能最基本的方法。镁锂合金中研究最多的是Al、Zn作为主要合金化元素的 Mg-Li-Al(LA)系、Mg-Li-Zn(LZ)系。它们除了Al和Zn的固溶强化外,还在时效过程中生成Li2Mg(Al,Zn)强化相产生时效强化;但Al、Zn含量大时合金强度虽高却脆性大,限制了LA、LZ合金向更高强度的发展[2-6]。因此,如何设计具有高温高强韧Mg-Li-M(M为合金化元素)合金是近年来镁锂合金研究的方向之一。已有研究结果表明,镁锂合金的相界面结合能力影响着材料的强度和韧性,对材料断裂方式的控制具有重要作用,尤其是界面微合金化对界面结合能力影响很大;但其在原子层次的微观机理还有待深入探讨。笔者尝试从界面能量学、微观电子结构角度探讨微合金化元素对α-Mg/β-Li界面的影响。
1 理论方法与模型
DVM方法是美国西北大学Ellis等人提出的在密度泛函框架下[7-8]数值求解KohnSham方程的一种计算方法。此方法最初主要用于量子化学的研究,后来逐渐扩展应用于金属、半导体、金属间化合物等固体领域的电子结构计算,获得了较好的结果[9-10]。DVM的主要思想是在位形空间(实空间)中选择一组分离的取样点,把单电子波函数(分子轨道)用一组数值原子基函数展开,然后代入Kohn-Sham方程,用其近似解确定误差函数,通过对误差函数中展开系数求变分,使得误差函数对所有取样点有极小值,得到久期方程,把微分方程变为代数方程。
计算采用CAST EP(Cambridge Serial Total Energy Package)软件包,选择LDA下的CA-PZ泛函描述交换能,平面波截止能均取为340eV,布里渊区的K点取为4×4×2,每个原子收敛精度控制在10-6eV。各原子的外层电子组态分别为:Li-2s1,Mg-3s2,Al-3s23p1,Zn-3s23p63d104s2,Si-3s23p2,Ca-3s23p63d2。首先对Mg、Li单胞结构进行优化,优化后晶格参数见表1所示。与实验数据相比,晶格参数a、b、c的误差均很小,因此本文采用的计算方法是可信的。图1所示为建立的Mg/Li界面模型,依据实验表征结果由Mg和Li的密排面组成,即Mg的(0001)面与Li的(011)面搭配构成,此模型由五层 Mg(0001)面和五层Li(011)构成。为了分析方便,笔者对界面附近的原子进行了编号。图1中微合金化元素(Zn,Al,Si,Ca)均为替位型杂质,分别替换模型中标号为1的Li原子和标号为2的Mg原子。在计算中,采用能量最小化方案对Mg与Li的层间距进行了优化。

表1 Mg、Li晶胞结构参数

图1 Mg/Li界面模型
2 结果与讨论
2.1 Mg/Li界面结合能力

图2 Mg/Li界面的拉伸示意图
Mg-Li二元合金中,α-Mg和β-Li是两个主要的相。前述分析了Mg、Li单胞的晶格参数,这里对图1中由α-Mg和β-Li构建的相界面结构进行单向拉伸模拟实验,Mg/Li界面经过拉伸之后的应力-应变曲线如图2所示。由图可以看出,在拉伸的初始阶段,应力随着应变的增加而增大;当应变增加到25%时,对应的应力达到最大6.3GPa。由于拉伸模拟用结构模型没有考虑缺陷,因此本计算所得抗拉强度远高于实际值,称为理想抗拉强度。当应力达到极限后,随着拉伸的继续,应力随着应变的增加而下降,直至体系最后断裂。总体来看,Mg/Li体系具有很大的延展性,当应变量达到80%时才断裂,表现出了良好的超塑性,与其实际中作为超塑性合金相符。

图3 Mg/Li界面应变分别为5%、7.5%、10%、25%对应的层间距变化
为了分析Mg/Li界面经过拉伸之后发生断裂的部位,图3显示出了拉伸过程中层间距的变化;Mg1、Mg2分别代表Mg基体部分中靠近界面处的第一层、第二层层间隙;Li1、Li2分别代表Li基体部分中靠近界面处的第一层、第二层层间隙;Mg/Li代表界面处的间隙。从图中可以看出,当体系的应变分别为5%、7.5%、10%时,各层的层间距也均匀增加;但当应变达到25%时,体系发生断裂,断裂部位为Mg/Li界面,表明 Mg/Li是整个体系中最薄弱的部分,只有提高Mg/Li界面的结合能力,才能有效改善Mg-Li合金的力学性能。
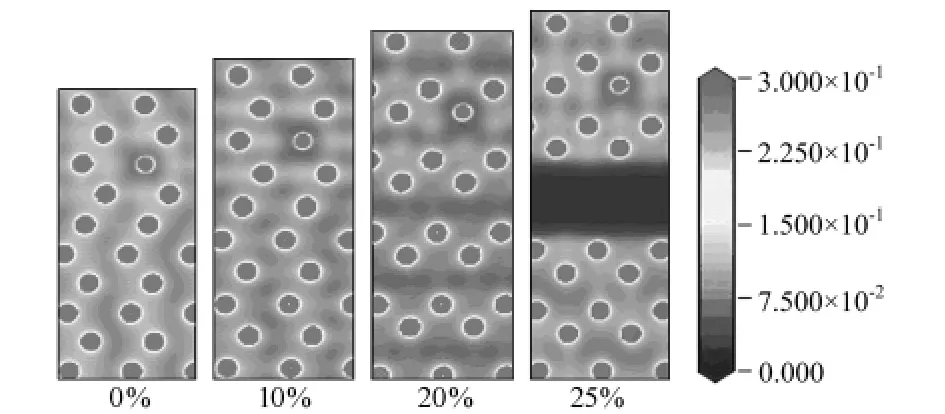
图4 应变分别为0%,10%,20%,25%时Mg/Li界面的电荷密度图
为了进一步分析不同拉伸阶段Mg/Li相界面电荷密度的变化,图4显示出了应变分别为0%,10%、20%、25%时的电荷密度。当体系没有施加载荷和应变量低于10%时,电荷密度分布均匀,说明体系应变量均匀,呈均匀变形;而当体系应变为20%、25%时,电荷密度以层为单位开始局域化,原子间的相对位置也发生了明显的变化,如断裂处电荷密度为零。
2.2 合金化对界面结合能力的影响
合金化是镁合金中改善其强韧性的主要方法,通常Al、Zn等是Mg-Li合金中最主要的合金化元素,均可起到固溶强化和析出强化的作用。另外,Si、Ca可起到析出细晶和强化的作用。无论何种元素,其在合金中所起的强化作用,均离不开与界面的联系,即合金化元素有在相界面、晶界面聚集的现象。下面将对Al、Zn、Ca、Si在 Mg/Li界面聚集产生的影响进行分析。
计算中,将Al、Zn、Ca、Si等分别替换图1中标号1的Li原子和标号2的Mg原子。研究发现,替换位置1和位置2后所得结果变化不大。由于实际中这些合金化元素主要是固溶到体心立方结构的Li中,故下面仅对这些合金化元素替换界面体心立方一侧Li(位置1)的计算结果进行分析,表2所示为计算结果。从表2可以看出,Zn偏聚于 Mg/Li相界面后的分离功大于纯 Mg/Li界面的值,表明Zn偏聚可增强界面间的结合力;而Al、Si的作用略弱于Zn的作用;相比较Ca在Mg/Li相界面偏聚后则会降低Mg/Li界面的结合力。各合金化元素偏聚于Mg/Li相界面后的分离功,即对界面结合能力的影响按以下顺序递减:Zn,Si,Al,Ca。由此可知,在合金设计中,Zn的晶界偏聚有利改善界面结合强度;Ca尽管可以细化晶粒,但应严格控制在合金体系中的含量,以避免在晶界析出,引起脆性。

表2 各元素偏聚于Mg/Li相界面后的物理参数
3 结论
1)理论计算表明,在拉伸力作用下,Mg-Li合金伸长率较高,表现出了较好的超塑性,这与实验结果相符;从拉伸断裂后Mg/Li界面断裂发生位置来看,Mg与Li形成的界面结合能力较弱,不利于超塑性的提高。
2)Zn、Al、Si、Ca富集于 Mg/Li界面时,对界面结合能力有不同程度的影响。其中,Zn有利于提高界面结合强度;Al、Si对界面结合能力影响不明显;Ca则会降低 Mg/Li界面的结合能力,不利于超塑性的改善。
[1]Gasior W,Moser Z,Zakulski W,et al.Thermodynamic studies and the phase diagram of the Li-Mg system[J].Metallurgical and Materials Transactions A,1996,27(9):2419-2428.
[2]Jackson J H,Frost P D,Loonam A C,et al.Magnesium-lithium base alloys-preparation,fabrication,and general characteristics[J].Journal of Metals,1949(2):149-168.
[3]Wu Libin,Cui Chongliang,Wu Ruizhi,et al.Effects of Ce-rich RE additions and heat treatment on the microstructure and tensile properties of Mg-Li-Al-Zn-based alloy[J].Materials Science and Engineering:A,2011,528(4-5):2174-2179.
[4]Alamo A,Banchik A D.Precipitation phenomena in the Mg-31at%Li-1at%Al alloy[J].Journal of Materials Science,1980,15:222-229.
[5]Busk R S,Leman D L,Casey J J.The properties of some magnesium-lithium alloys containing aluminum and zinc[J].Journal of Metals,1950(7):945-951.
[6]Jones W R D,The mechanical properties of binary and ternary magnesium alloys containing lithium [J].Journal of Institute of Metals,1955-56,84:364-378.
[7]Hohenberg P,Kohn W.Inhomogeneous electron gas[J].Phys Rev B,1964,136:864.
[8]Kohn W,Sham L J.Self-Consistent Equations Including Exchange and Correlation Effects[J].Phys Rev A,1965,140:1133.
[9]Wang F H,Wang C Y.First-principles investigation of hydrogen embrittlement in polycrystalline Ni3Al[J].Phys Rev B,1998,57:289.
[10]王福合,杨金龙,李家明.Al(111)表面单个 Al原子的操纵钨针尖与 Al原子的相互作用[J].物理学报,1998,47:1827.
[11]Alamo A,Banchik A D,Precipitation phenomena in the Mg-31at%Li-1at%Al alloy[J].J Mater Sci,1980,15(1):222-229.
[12]Levinson D W,Mcpherson D J.Phase relations in magnesium lithium aluminum alloy[J].Trans Am Soc Met,1956,48:689-696.
