应用ABEEM/MM研究Al(Ⅲ)离子水分子体系
2012-09-15宫利东
彭 楠,宫利东
(辽宁师范大学 化学化工学院, 辽宁 大连 116029)
应用ABEEM/MM研究Al(Ⅲ)离子水分子体系
彭 楠,宫利东
(辽宁师范大学 化学化工学院, 辽宁 大连 116029)
采用高水平的从头算方法,对铝离子水分子团簇 Al3+-(H2O)n(n=1-6) 在 MP2/6-31++G(d,p)水平下进行了结构的研究。采用了BSSE校正且在MP2/6-311++G(2d,2p)水平下对能量进行了计算,并运用原子-键电负性均衡浮动电荷模型(ABEEM/MM),确定了Al3+-H2O的参数,将Al3+-H2O参数应用于Al3+-(H2O)n(n=1-6)体系中,计算内容包括结合能和连续结合能等,所得结果和从头算的结果符合较好。
铝离子水分子团簇;ABEEM/MM模型;结构优化计算
目前,普遍所采用的研究水合离子方法有X射线散射(XD)、核磁共振 (NMR)、AIMD[1]、分子动力学模拟(MD)等。然而,这些方法普遍耗费大量的时间和资源,因此需要采用便捷的、低能耗的方法势在必行。根据电负性均衡方法, 本课题组建立了原子-键电负性均衡方法(ABEEM)[2,3]可用来简单进行计算。
本文采用从头算方法,对 Al3+-(H2O)n(n=1~6)的结构、结合能,连续结合能等性质进行了计算,构建了Al3+-(H2O)n之间的精确势能函数。本论文所采用的模拟方法在计算上既节省了大量研究时间精力,也节省了资源,因此工作效率有很大的提高,其结果与其他模拟方法大体一致。
1 计算模拟方法
1.1 理论方法
本文用MP2方法,从头计算采用Gaussian03[4]程序完成,在MP2/6-311++G(d,p)//MP2/6-31++G (2d,2p)水平上进行计算。
1.2 ABEEM/MM理论模型建立

Eb表示键长的伸缩振动势能用Morse势能函数表示:
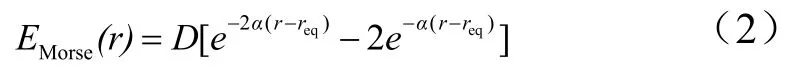
在ABEEM-7P[5]力场中用谐振势来表示:

θ为水分子中H-O-H键角,θeq为H-O-H的平衡键角,fθ为角力常数,Evdw表示van der Waals相互作用能,Eelec表示静电作用能。
总的能量表达式见表达式(4):
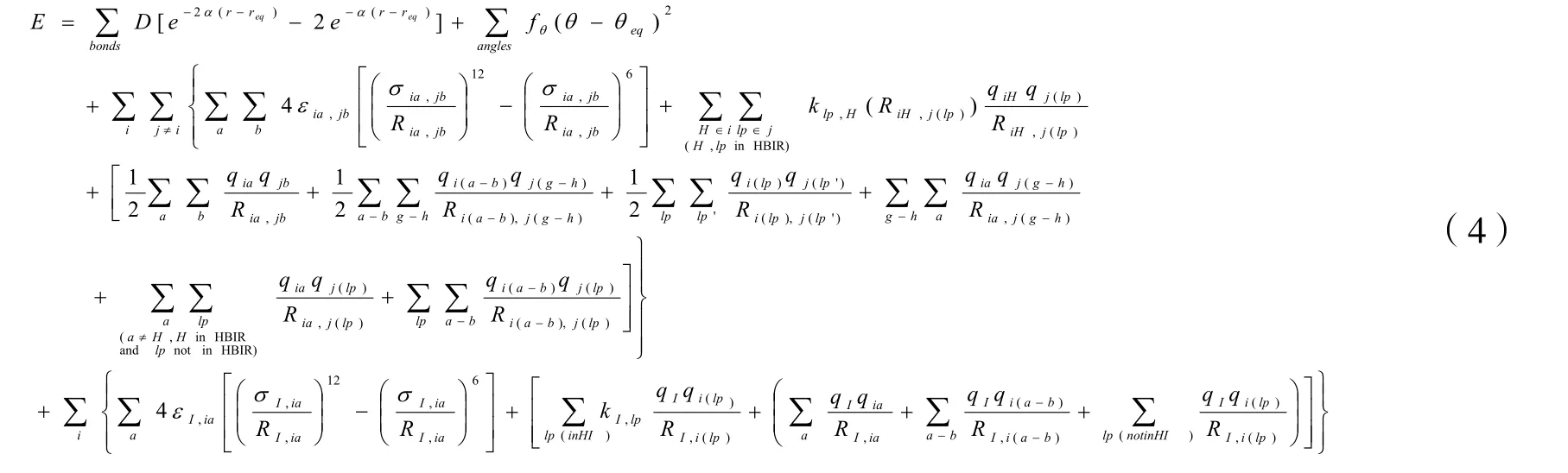
公式(4)中,最后一项为Al3+与水分子之间的相互作用能,本文拟合的Al3+的具体关系为:

2 结果与讨论
2.1 结构优化
从图1可以清楚得到1到6个铝离子水分子团簇的结构和对称性情况,它们都是低能构型,并且能较稳定存在于水溶液中,可以为我们呈现空间立体效果,是研究结合能,连续结合能最基本的构型。
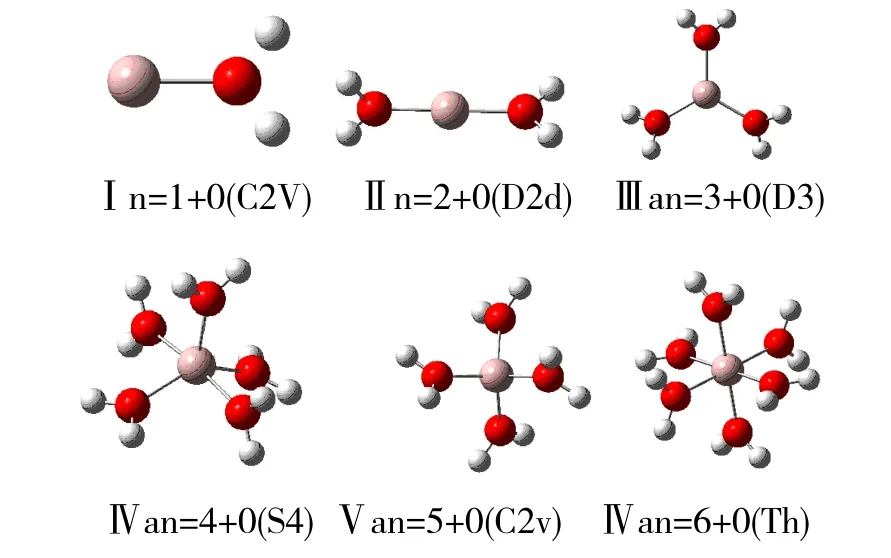
图1 Al3+(H2O)n(n=1-6)的低能集合构型(对称性)Fig.1 The low-energy conformations of Al3+(H2O)n(n=1-6)(symmetry)
从图2(A),图2(B)可得出计算得到的键长,结合能随n的变化都成上升趋势,并且它们的变化相对明显,而连续结合能由图2(C)可看出逐渐下降,但结果都和从头算大体一致,这说明所采取的方法是合理的。
3 结束语
利用从头算方法,将原子-键电负性均衡方法融进分子力场ABEEM/MM 模型中建立金属离子水分子模型,将该模型应用到金属铝离子水分子团簇中,通过参数确定其势能函数,计算出铝离子水分子团簇的结构,当n从1增加到6, 键长逐渐变长。
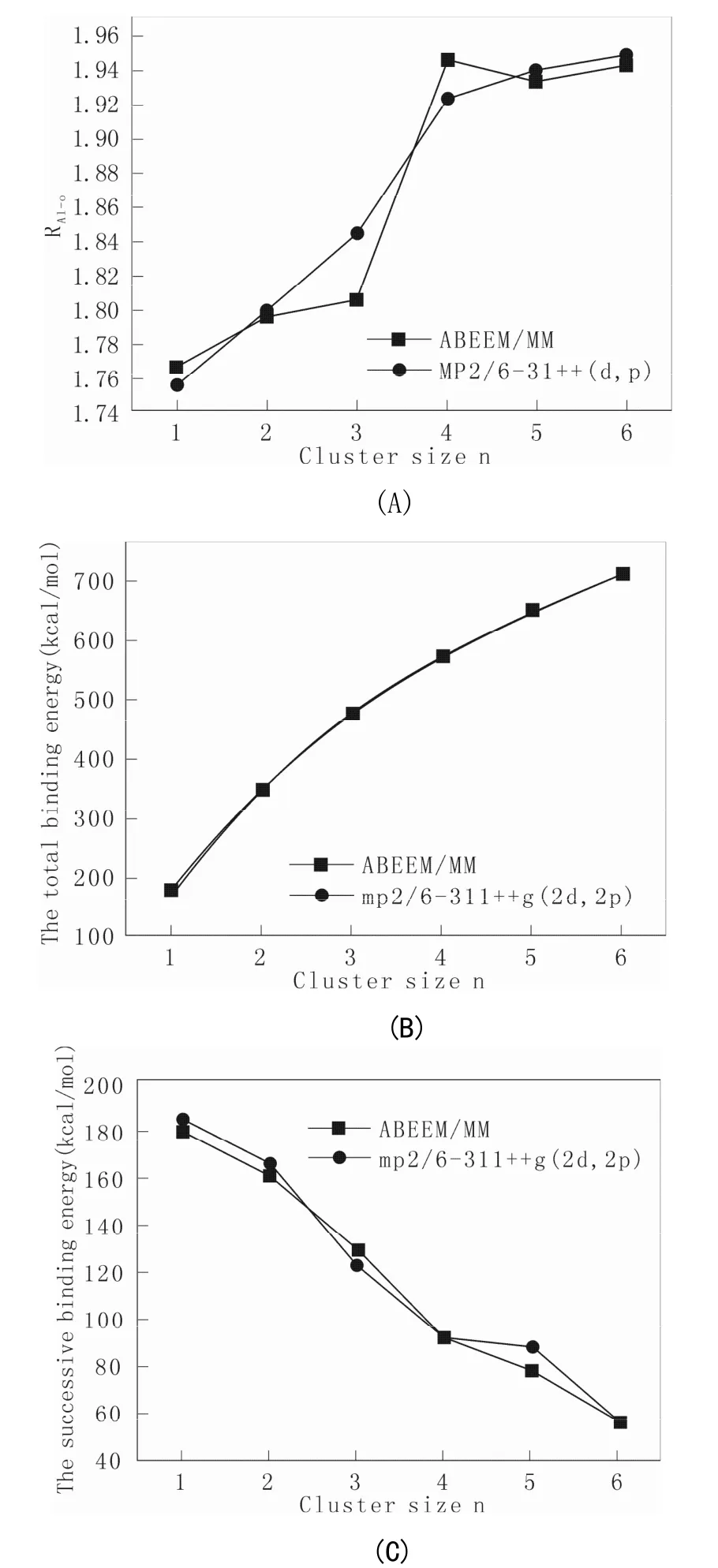
图2 Al3+(H2O)n(n=1-6)的键长RAI-O(A),总的结合能△En(B)和总的连续结合能△En-1(C)与n的变化曲线Fig.2 The RAl-O, total binding-energy △Enand successive binding-energy △En-1of Al3+(H2O)n(n=1~6) curve with n

表1 Al3+(H2O)n(n=1-6)的键长RAI-O、总的结合能△En,总的连续结合能△En-1Table 1 The RAl-O, total binding-energy △Enand successive binding-energy △En-1of Al3+(H2O)n (n=1~6)
这是由于随 n的增加, Al3+与每个水分子的平均相互作用在逐渐减弱,对于结合能随着 n的增加,逐渐均匀地增大而对于连续结合能刚开始变化较快,随后比较缓慢,总的趋势是逐渐减小,并且ABEEM/MM计算的键长RAl-O、结合能△En和连续结合能△En-1相对于从头算计算结果的平均绝对偏差分别为 0.001 2 Å、2.8 kcal·mol-1和3.6 kcal·mol-1,所得结果和从头算对比都取得了较好的结果,该文章为其它金属离子水团簇的研究提供了一定的参考价值。
[1] Masia M. Ab initio based polarizable force field parametrization[J]. J Chem Phys, 2008, 128: 184107.
[2] Yang Z Z, Wang C S. Atom-bond electronegativity equalization method.Ⅰ.Calculation of the charge distribution in large molecules[J]. J. Phys. Chem. A, 1997, 101: 6315-6321.
[3] Wang C S, Yang Z Z. Atom-bond electronegativity equalization method.Ⅱ.one-pair electron model[J]. J. Chem.Phys., 1999, 110: 6189-6197.[4] Frisch M J, Trucks G W, Schlegel H B, et al. Gaussian 03, Revision D 01[M]. Wallingford, CT: Gaussian Inc, 2004.
[5] Wu Y, Yang Z Z. Atom-bond electronegativity equalization method fused into molecular mechanics. Ⅱ. A seven-site fluctuating charge and flexible body water potential function for liquid water[J]. J Phys Chem A, 2004, 108: 7563-7576.
ABEEM/MM Simulation of Aluminum (Ⅲ) Cation Water Systems
PENG Nan,GONG Li-dong
(Liaoning Normal University, Liaoning Dalian 116029,China)
An ab initio method with high level was used to study aluminum ionic clusters Al3+-H2On(n=1~6) at the MP2/6-31++G(d,p) level. The energy was exactly calculated with BSSE correction at the MP2/6-311++G(2d,2p) level. Parameters of Al3+-H2O was determined with atom-bond electronegatibity equalization fluctuating charge mode (ABEEM/MM). The parameters were used in the study of aluminum (Ⅲ ) cation water clusters Al3+-H2On(n=1-6),including binding-energy and successive binding-energy. The results are well consistent with ab initio method.
Metal ion water clusters;ABEEM/MM model; Geometry optimization calculation
TQ 133
A
1671-0460(2012)09-0922-03
2012-03-24
彭楠(1986-),女,辽宁沈阳人,硕士,2012年毕业于辽宁师范大学物理化学专业,研究方向:理论与计算。E-mail:bsbs9494@163.com。
