有监督的主成分分析和偏Cox回归模型在基因数据生存预测中的应用*
2012-03-11覃婷王彤
覃 婷 王 彤
当用基因表达数据预测生存情况时,基因数远远超过了样本例数。除了高维度以外,基因表达之间通常存在着某种未知的相关,其增加了解释变量之间的共线性。基因表达数据存在的小样本、高维度、强相关的特点给生存预测带来了困难。因此根据基因数据做生存预测时,首先需要对基因表达数据进行降维或者调整,从而更加有效而准确的进行参数估计。有监督的主成分分析(supervised principal component analysis,SuperPC)和偏 Cox回归(partial least squares Cox regression)是其中的两种降维方法。本文将通过模拟研究和对国际上公开的三个基因数据集进行分析,以探讨这两种方法用于高维数据生存预测表现的优劣,为得到更精确的预后估计和改进治疗策略提供依据。
基本原理与方法
1.基因微阵列数据的标识与比例风险模型
假设有一组包含着截尾数据的基因微阵列生存数据有 n 个个体,(yi,δi,xi);i=1,…,n。其中 yi为个体i的失效时间,yi可以是完全数据,也可是截尾数据;δi是一个指示变量,当δi=1时为完全数据,而当δi=0为截尾数据;xi=(xi1,…,xip)T为个体i的自变量向量。
令Y为生存时间。生存函数定义为S(y)=P(Y>y),是某个体在时间y时刻依然存活的概率。风险函数测量在y时刻存活的个体,在下个很小的时间段内死亡的瞬时风险。比例风险模型表示为
h(y,X)=h0(y)exp(XTβ) (1)其中h0(y)是一个非指定的基准风险函数。
模型的参数向量^β通常可以取最大偏对数似然得到,基准生存函数H0(y)可用Breslow估计,表示为 ^H0(y)。对于一个表达谱为~X的新样本,根据已知的参数和基线风险估计求出其风险函数和生存函数,
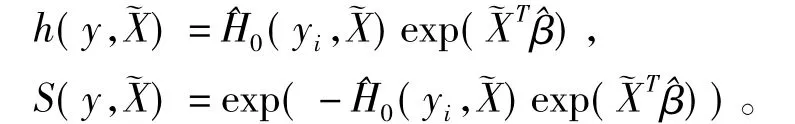
2.有监督的主成分分析
SuperPC 是由 Bair和 Tibshirani等人提出的〔1-2〕,它改进常规主成分分析无法保证所选择的主成分与病人的生存相关的缺点,在降维的时候考虑了生存时间,其核心思想就是只对与生存时间密切相关的基因进行主成分分析。
该方法首先将每一个基因分别代入单变量Cox模型h(t|x)=h0(t)exp(βx),以检验它们对生存的影响。然后对其进行基于偏似然函数的参数估计与假设检验,检验方法为似然比检验。将基因按照检验所得到的P值从小到大排序,然后根据交叉验证法挑选出前λ1百分比的基因组成一个简化矩阵Xθ。采用奇异值分解法(singular value decomposition,SVD)对这个简化矩阵进行主成分分析。
假设X矩阵的列已经被中心化,均数为0。那么n×p矩阵X的奇异值分解写作:

其中U是一个n×n的正交阵,V是p×p正交阵,D是一个以奇异值dj为对角元素的n×p对角阵,r=min(n,p)是X的秩,非零奇异值的数目与X矩阵的秩相等,d1≥d2≥…≥dr>0。
那么,简化矩阵Xθ的奇异值分解写作:

令 Uθ=(uθ,1,uθ,2,…,uθ,r),称 uθ,1为 X 的第一有监督的主成分,依此类推。如果仅取一个成分,即拟合一个应变量为y和自变量为uθ,1的Cox比例风险模型,得
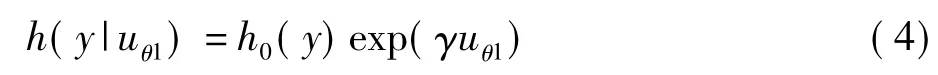
从公式(3)变换得到(注意到正交阵V'V=I),

因为 uθ,1是 Xθ的一个线性组合:uθ,1=Xθwθ,1,所以模型(4)可以看作是一个利用了Xθ中的所有自变量的受限模型:
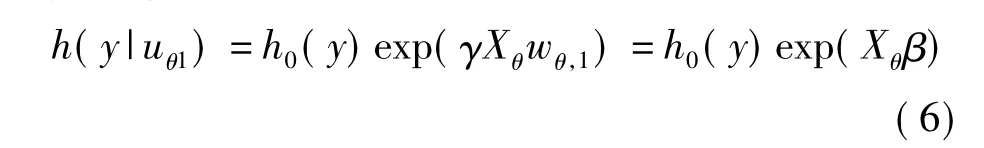
假如有一个新的基因数据集x*,对其进行生存预测,步骤如下:

3.偏Cox回归
在基因数据的生存预测方法中,基于偏最小二乘的生存分析是一个重要的家系。利用偏最小二乘方法进行降维,即从原始变量中提取偏最小二乘成分,然后将提取的这些线性成分应用于标准的Cox回归进行生存预测的方法,就称之为偏Cox回归。
偏Cox回归算法有很多种,这里采用的是Nygård提出的算法〔3〕。该算法主要是通过将生存问题转换为广义线性回归问题,然后依照广义线性模型的迭代再加权偏最小二乘算法提取PLS成分,从而实现高维数据的降维,然后将所得到的参数估计以及提取的PLS成分代入Cox比例风险模型中,进行生存预测。由于这种算法将基准风险增量的估计与PLS降维分开,使得PLS的成分仅为基因表达谱的线性组合,更符合生物解释。
由于该算法只是对^η进行了部分更新,可解决收敛速度过慢的问题。且分开估计协方差效应和基线风险增量避免了数据维度的扩张,加上提取的PLS成分数量小,所以计算速度通常很快,节约了计算时间。
4.根据交叉验证法选择模型调整参数
预测方法的模型复杂程度是由估计调整参数来决定的。调整参数的估计方法有很多种,最经常使用的就是交叉验证。在本文中,我们采用Verweij和van-Houwelingen提出的交叉验证准则〔4〕,这种准则是建立在Cox偏对数似然的基础上的。
首先将数据分成等大小的K个部分(1<K≤n),每个第i(1≤i≤K)次的交叉验证都会将第i个层剔除,只用剩下的层来训练模型,根据训练好的模型来估计被剔除的第i个层的预测信息,重复K次,这样每一个部分都做了并且只做了一次验证组。令l(β)表示全部数据的Cox对数似然,l(-i)(β)表示剔除第i个层数据的对数似然,将第i个部分对似然的贡献定义为li(β)=l(β) - l(-i)(β),使得 l(-i)(β)最大化的 β 估计值表示为β^(-i)。假设似然成分是独立的,那么li(β)就简单的等于第i个部分的贡献,并且l(β),K折交叉验证的最大对数似然为 CVL =,随着λ的变化,每个最大对数似然CVL也在变化,其中最大的CVL所对应的λ为最优调整参数。
在实践中,通常会给出调整参数的范围,在给定的范围内找最优调整参数。SuperPC的调整参数λ=(λ1,λ2)是双变量的,λ1代表单变量分析中与生存时间有关的基因子集占基因全集的百分比,λ2代表将要选择的主成分数的范围。
5.模型评价
一般来说,预报因子的真正性能评价应该用一个独立的数据来完成。但是在缺乏独立数据的情况下,可以通过交叉验证来进行模型评价,即将每一个数据集按2:1的比例随机分成训练数据集和验证集。训练组样本的基因表达和生存数据被用来构建预测模型,验证集用来评估模型的性能。为了避免依赖于训练集和验证集的选择,需进行重复分组,以所有评价标准的结果的中位数和四分位数间距来估计预测模型的性能。本论文主要的评价标准为决定系数R2。
决定系数是验证组中的生存数据可以被预报因子解释的那部分变异所占的百分比。预测性能良好的预报因子可以解释验证组生存数据的绝大部分变异。在传统的回归背景下,R2=1-残差平方和/总平方和,因此它的取值范围在0到1之间。然而这个定义在数据存在删失的情况下不能使用,因此,Nagelkerke给出了一个可以用在Cox比例风险模型中R2统计量〔5〕
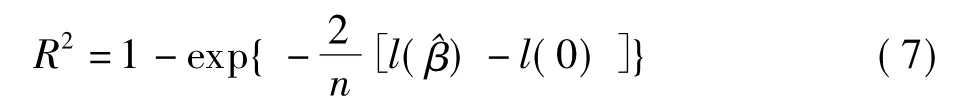
其中l(.)表示对数似然函数。R2越大,预报器的预测性能越好。
模拟分析
为了进一步验证上述模型的预测性能,我们根据基因数据的特点设计了模拟实验,用上述方法对模拟实验产生的数据进行分析,根据模型预测评价标准对它们的预测性能进行比较。模拟条件设定如下:
(1)协变量矩阵X:
生成100×1000的基因协变量矩阵,每一行表示一个病人,每一列表示一个基因。这些协变量服从多元正态分布,均数向量为0。将数据分成十块等大小的基因块∑b,令它们的方差协方差矩阵的对角元素为,非对角元素为。因此∑对应于基因表达的类别,这样不同类别的基因表达是独立的,但是在同一个类别中的基因表达有同样的两两相关。在模拟中,我们令ρ分别等于0.3,0.6,0.9以观察不同相关程度对结果的影响,同时评判三种不同方案的方差:(a)=…==1,即所有的基因表达的方差相等,(b)==2,=…==1,即前两块的基因表达的变异更大,(c)==1/2=…==1,即前两块的基因变异比其他块的要少。
我们首先产生服从(0,1)均匀分布的随机数S,令生存函数S(t)=S,利用用产生相应的生存时间t。
(4)生成删失指示变量:
产生一组随机数,服从二项分布,发生1的概率为0.8,也就是截尾为20%,为了观察截尾比例是否对降维方法产生影响,我们改变了截尾比例,截尾比例分别为20%,50%。
按照上述的实验设计产生训练组数据,然后以同样的条件产生验证组数据。分别应用SuperPC和偏
(2)参数的设定:
每个回归系数对应于它对应变量的影响。在本文中,参数的设定如下:当 1≤j≤100,βj=0.01,当 101≤j≤200,βj从0.01 到0.1,每0.01 为一个步长。201≤j≤1000,βj=0,表示在基因矩阵中只有少数一些相关的协变量,大部分的协变量都是无关的。
(3)生成生存时间:Cox回归方法对训练组进行建模,然后用验证组数据进行预测评价,在最优调整参数的条件下评价模型的预测性能,评价标准为R2,从评价标准的中位数以及离散程度来对模型进行评价。以上过程重复200次。模拟结果以箱式图矩阵表示。
箱式图矩阵的行代表影响生存的前两块基因的方差,列代表3个不同的相关系数。spcr为SuperPC方法,pls为偏Cox回归方法。censored=0.2和censored=0.5分别表示删失比例为20%和50%的模拟数据的结果。
由图1可见,根据模型评价标准,SuperPC的预测性要优于偏Cox;当相关系数相同时,两种方法得到的R2都是随着方差的增大而增大,提示基因的方差越大的时候,基因块越容易被识别。方差相同时,相关系数越大,得到的R2越大。随着截尾比例的增大,模型的预测性能会变差,说明模型的预测能力会受到删失比例的影响。
实例分析
用上述两种方法对国际上三个公开的基因数据集进行了分析,它们分别是Van't Veer等的乳腺癌数据〔6〕,78个病人,4 751个基因,观察事件的结局是乳腺癌是否转移,截尾比例为56.4%;Beer等人的肺癌数据集〔7〕有86个病人和7 129个基因,观察事件结局为死亡,截尾比例为72.6%;Bullinger等人急性髓系白血病(acute myeloid leukemia)的数据〔8〕,116 个病人,6 283个基因,观察事件结局为死亡,截尾比例为42.24%。
因为对于每个数据集,采用不同的评价标准,最佳的预测方法有可能是不同的〔9〕。因此,对于一个实例数据,首先要看用这两种方法进行分析的预测性能如何,然后根据预测结果挑选合适的预测模型。
首先按2:1的比例将数据随机分成训练组和验证组:训练组用于构造模型,而验证组用来对模型的预测性能进行评价。为了保证预测结果评价的客观性,避免数据任意分割导致的预测偏差,按上述方法重复将数据集随机分割200次。结果见图2。
图中bc代表乳腺癌数据,lc代表肺癌数据,aml代表急性髓系白血病数据,spcr为是SuperPC方法的分析结果,pls为偏Cox方法的分析结果。
R2值越大,模型可以解释数据的变异部分越大,模型的预测性能越好。从图2中我们可以看到,对于乳腺癌数据,SuperPC方法的表现要优于偏Cox回归。而对于肺癌数据和急性髓系白血病数据,则偏Cox回归的表现要优于SuperPC方法。
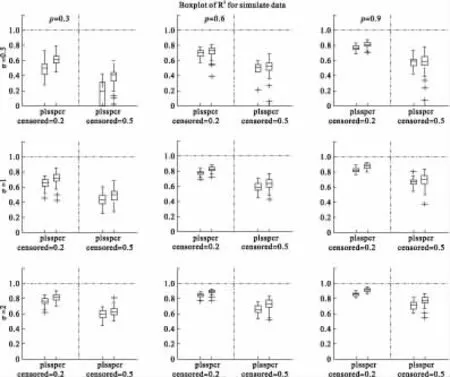
图1 模拟数据分析结果矩阵图
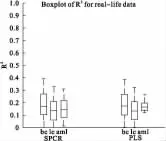
图2 实际数据分析结果图
因为我们对这3个数据进行了随机分割(分割成训练集和验证集),分割了200次,每次分割得到的最优调整参数的取值是不一样的。因此,这里我们给出了三个实例数据分析中,两种方法各自所选择的最优调整参数的分位数的表。
从表1中我们可以看到,SuperPC提取的成分数大于等于偏Cox回归,并且提取的成分数不稳定。SuperPC提取的基因子集占原基因集的比例很小,即简化矩阵远小于原始矩阵。所以虽然SuperPC引入的成分数多,但每个成分中包含的自变量数目要少于全基因集。偏Cox方法提取的成分数稳定,用很少的成分就可以解释原始变量的大部分变异。
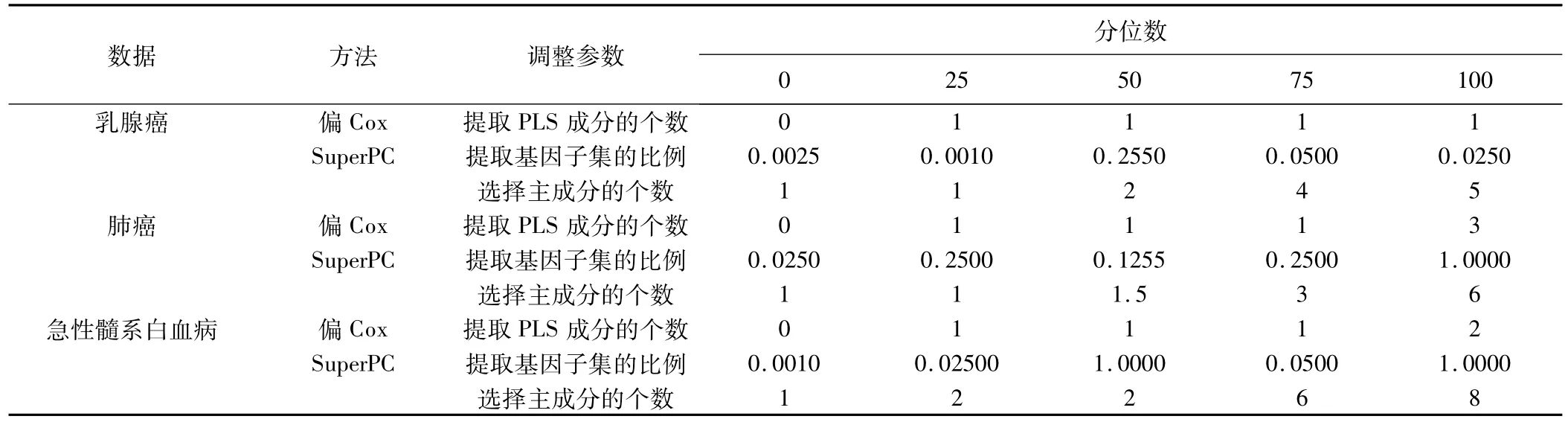
表1 实际数据分析中选择的最优调整参数的分位数
根据上面的模型预测性能评价,挑选各个数据对应的最适方法来对整个数据集进行了模型拟合就得到了预测方程。可以计算预后指数来估计病人的预后情况,预后指数的公式为PI=X^β。当得到新的病人的基因数据~X的时候,就可以根据预后指数PI=~X^β来对其分类:按照预后指数的中位数,将病人分成两组,预后指数超过中位数的,为高危组,可考虑相对积极的治疗策略;低于中位数的,为低危组,可以采用相对保守的治疗方案。
讨 论
有监督的主成分分析和偏Cox回归都是将Cox比例风险模型与降维技术结合起来,适用于基因数据的生存预测。这两种方法都对主成分回归进行了改进,利用了应变量的信息。它们的区别在于有监督的主成分分析是对基因子集进行特征提取,而偏Cox回归则是对基因的全集进行特征提取。
从模拟实验中可见:(1)随着影响生存的基因块的方差的增大,两种方法的预测性能变好,这是因为当自变量的变异更大的时候,被提取的信息也更多,可提高回归估计的精度和稳定性,故而当影响生存的基因块的方差大的时候较容易被识别出来。(2)随着组内相关系数ρ的增高,两种方法的预测性能都变好。因为只有存在着相关,才能够把维度降下来,特征值会随着相关的增高而增大,那么前几个主成分的方差就会很大,这样只需要几个主成分,就能够很好的解释原始变量的变异。随组内相关系数的增高,预测方法性能变好的同时还应注意到,当基因的相关度较高的时候,预测方法对选择正确的基因比较不敏感,对生存没有影响却与对生存有影响的基因高度相关的基因也可能被纳入。(3)随着删失比例的增加,两种方法的预测性能变差,说明预测方法会受到删失比例的影响。
在实例分析中,根据判断标准,不同的数据集最优预测方法不同。模拟研究和实例分析中,有监督的主成分分析提取的成分数要大于等于偏Cox回归。然而我们也应该注意到,因为这种主成分是有监督的,所以虽然引入的成分数多,但每个成分中包含的自变量数目要少于全基因集。
在我们的模拟研究中,这两种方法的预测性能很好,用我们的模型能够解释原始变量的变异比例较大,决定系数甚至达到了0.9的情况。但是,实例分析中可以看到,决定系数没有那么大。造成这种差异的原因可能是因为模拟实验设计的时候,基因块之间设定了相关系数,各个基因块中的基因两两相关,而基因块之间不相关,并且对回归参数也进行了设定,只是前两块基因与生存有关,数据生成具有一定的规律。而实际的微阵列数据远比模拟数据要复杂得多,基因之间以未知的方式相关,并且微阵列数据中的协变量数千至数万个,简单的模拟不能够捕获这种复杂的关系。并且模拟中为了计算的方便,只是模拟了1 000个协变量,样本含量固定取100个,这与实际微阵列数据的样本含量和自变量个数相比,样本含量的比例要大得多。实例数据分析,因为是随机拆分数据,最后得到的训练组数据和验证组数据的截尾比例可能会比原来的数据要高,这也是影响结果的一个原因之一。
在模拟研究中,有监督的主成分分析的预测性能要优于偏Cox回归。但本文介绍的偏Cox回归算法因其在收敛性上进行了改进,从而计算速度很快,比有监督的主成分分析方法节约了很多时间。对于高维数据的分析来说,计算时间短也是个很重要的优点。
1.Bair E,Tibshirani R.Semi-supervised methods to predict patient survival from gene expression data.PLoS Biology,2004,2:511-522.
2.Bair E,Hastie T,Paul D,et al.Prediction by supervised principal components.Journal of the American Statistical Association,2006,101:119-137.
3.Nygård S,Borgan O,Lingiaerde OC,et al.Partial least squares Cox regression for genome-wide data.Lifetime Data Anal,2008,14:179-195.
4.Verweij PJMvan,Houwelingen HC,Cross-validation in survival analysis.Stat Med,1993,12:2305-2314.
5.Nagelkerke NJS.A note on a general definition of the coefficient of determination.Biometrika,1991,78:691-692.
6.Van't Veer LJ,Dai H,Van de Vijver.Gene expression profiling predicts clinical outcome of breast cancer.Nature,2002,415:530-536.
7.Beer DG,Kardia SL,Huang CC,et al.Gene-expression profiles predict survival of patients with lung adenocarcinoma,2002,Nat Med 8:816-824.8.Lars Bullinger MD,Konstanze Döhner MD,Eric Bair,et al.Use of geneexpression profiling to identify prognostic subclasses in adult acute myeloid leukemia.Massachusetts Medical Society,2004,350 16:1605-1616.
9.W.van Wieringen,D.Kun,R.Hampel,et al.Survival prediction using gene expression data:a review and comparison.Computational Statistics and Data Analysis,2009,53:1590-1603.
