医用HMDI基聚碳酸酯型聚氨酯弹性体的合成与表征
2012-02-15姜秀娟余成科陈大福张立群
姜秀娟,石 锐,余成科,陈大福,张立群,2*
(1.北京化工大学有机无机复合材料国家重点实验室,北京100029;2.北京化工大学北京市新型高分子材料制备与成型加工重点实验室,北京100029;3.北京市创伤骨科研究所,北京100035)
0 前言
聚氨酯弹性体具有良好的生物相容性、力学性能及耐久性、耐磨性等,现已广泛应用于心脏起搏器绝缘线、人工血管、介入导管、人工关节、人工软骨、神经导管、控制释放载体等生物医学领域中1-2。
医用聚氨酯弹性体按多元醇种类可分为聚酯型、聚醚型和聚碳酸酯型等。聚酯型酯软段易水解,且在体内酶作用下加速水解[3-4],因此不适合做长期植入材料。目前应用最为广泛的是聚醚型,但其易在血液中巨噬细胞作用下氧化降解或金属离子诱导降解,导致生理条件下的应力开裂,造成性能稳定性下降[5-6]。目前生 物 稳 定 性 最 好 的 是 聚 碳 酸 酯 型[7-8]。Szycher等[9-10]研究表明,聚醚型聚氨酯可在体内较长时间稳定存在,但植入体会产生环境应力微裂纹,且微裂纹与醚键多少有关;这些微裂纹不仅降低聚合物性能,而且会成为血栓成核点,最终导致失效破坏;聚碳酸酯型聚氨酯可以克服微裂纹问题。但研究表明其抗水解、抗酶解能力还有待提高[11-12]。
在异氰酸酯中,二苯基甲烷二异氰酸酯(MDI)是用途最为广泛的一种原料。但已有研究表明MDI在体内降解会产生可强烈致癌、可诱导基因突变物质4,4′-二苯甲烷二胺(MDA)[13-14]。
基于上述两方面综合考虑,要制备一种可长期植入体内的生物稳定性弹性体,非MDI基聚碳酸酯型聚氨酯弹性体为优选配方。Tang等[15]用聚六亚甲基碳酸酯二醇、六亚甲基二异氰酸酯(HDI)及BDO合成了不同硬段含量的聚氨酯,并研究了其酶催化水解稳定性,表明硬段含量、软段结晶性及氢键化程度对水解稳定性都有一定的影响。薛燕等[16]用溶液预聚法合成了以聚1,6-己二醇碳酸酯二醇(PHMCD)、异佛尔酮二异氰酸酯(IPDI)和三羟甲基丙烷(TMP)为原料的聚氨酯,研究了原料配比对其力学和形状记忆性能的影响。而目前基于HMDI的聚碳酸酯型聚氨酯少见报道。
HMDI在化学结构上与MDI相似,用环己基六元环取代苯环,属脂环族二异氰酸酯,可制得有优异光稳定性、耐候性和力学性能的不黄变聚氨酯制品[17]。The rmomedics公司开发了以HMDI为硬段结构的聚醚型聚氨酯材料 Tec oflex(tm)。Szycher等[13]研究证明由于Tec oflex(tm)不含苯环结构,因而不会产生致癌物质MDA。
本文采用脂环族异氰酸酯HMDI,合成了一种医用聚碳酸酯基透明聚氨酯弹性体,表征了其微相分离结构、力学性能、水解性能及细胞毒性等,为制备一种可长期植入体内的生物稳定型医用材料奠定基础。
1 实验部分
1.1 主要原料
PCD,相对分子质量2000,工业级,日本聚氨酯工业株式会社;
HMDI,化学纯,德国Bayer公司;
BDO,分析纯,天津市博迪化工有限公司;
BiCAT®8118,分析纯,美国Shepchem公司;
磷酸盐缓冲液(PBS,p H=7.2~7.4),分析纯,北京中衫金桥生物技术有限公司。
1.2 主要设备及仪器
傅里叶变换红外光谱仪(FT-IR),Tensor 27,德国Bruker Optik公司;
差示扫描量热仪(DSC),STARe system,瑞士Mettler-Toled公司;
动态力学热分析仪(DMTA),VA3000,法国01dBMetravib公司;
凝胶渗透色谱仪(GPC),Waters150-C,美国 Waters公司;
万能材料试验机,CMT5000,深圳新三思公司。
1.3 样品制备
预聚物的合成:将计量好的PCD在三口烧瓶中120~160℃真空脱水2~4 h,降温至50~70℃,加入HMDI,于70~80℃恒温反应2~3 h,脱泡得到预聚物A组分;将计量好的BDO与BiCAT混合30 min,脱泡得到B组分;
弹性体的制备:将计量好的A组分与B组分在三口烧瓶中80~90℃脱泡、搅拌至黏度较大但尚未凝胶时,迅速将物料倒入模具中浇注成型,100℃固化24 h,脱模、室温存放7 d后进行性能测试。合成的1#~5#聚氨酯样品的硬段含量分别为28.8%、29.2%、32.5%、35.7%、38.9%。
1.4 性能测试与结构表征
化学结构表征:采用傅里叶变换红外光谱仪表征聚氨酯的化学结构,ATR法进行测试;
热性能测试:采用差示扫描量热仪测试聚氨酯的玻璃化转变温度(Tg)及结晶行为,N2气氛,以20℃/min的速率从25℃升温至200℃保温5 min后,以10℃/min的速率降温至-100℃,然后以10℃/min的速率升温至200℃;
微相分离结构表征:采用ATR法傅里叶变换红外光谱仪及动态力学热分析仪表征聚氨酯的微相分离结构,测试频率1 Hz,应变0.1%,升温速率3℃/min,升温范围为-100~150℃;
相对分子质量及分散系数测试:采用凝胶渗透色谱仪测试聚氨酯的相对分子质量及分散系数,溶剂为四氢呋喃,聚苯乙烯为标样,进样体积为50μL;
按照GB/T 528—1998采用万能材料试验机进行拉伸性能测试,拉伸速率为50 mm/min;
按照GB/T 7757—2009采用万能材料试验机进行压缩性能测试,A法;
水解性能测试:样品放置在37℃的磷酸盐缓冲液中,每3 d换一次液,维持p H不变,分别浸泡1、3、5、7、10、20、30、40、70、100 d,用精度为0.0001 g的机械天平称量,计算质量损失率及吸水率,取3个样品的平均值;
细胞毒性测试:按照GB/T 16886.5—2003将材料浸提液与小鼠成纤维细胞(L929)接触培养,进行了细胞形态学观察,并用3-4,5二甲基-2-噻唑-2,5-二苯基溴化四唑(MTT)比色法对细胞毒性进行评价。
2 结果与讨论
2.1 红外分析
PCD与3#的红外谱图如图1所示,因1#~5#的红外谱图相一致,为清晰起见,本文中省略。从图1可以看出,PCD在3546 cm-1附近有明显的—OH特征吸收峰,而在合成的聚氨酯中此吸收峰消失,说明PCD的羟基全部参与了反应;聚氨酯在2273~2242 cm-1处无—NCO吸收峰,说明制品无—NCO残留。与PCD相比,聚氨酯中1740 cm-1附近自由峰形变宽,分裂出1710 cm-1附近键合特征峰;出现了3360 cm-1附近键合—NH伸缩振动峰以及1525 cm-1处C—N—H面内对称弯曲振动峰。由此证明合成的材料具有聚碳酸酯型聚氨酯的结构。

图1 原料PCD及合成的聚氨酯弹性体的红外谱图Fig.1 FT-IR spectra for PCD and synthetic PU elastomer

式中A峰1——位于分峰范围内峰1曲线,峰位一般在1740 cm-1左右,为自由峰
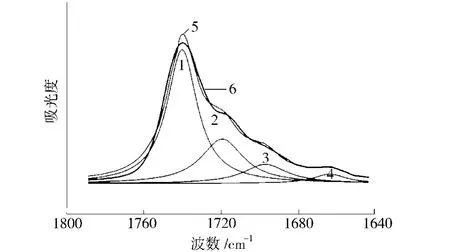
图2 红外谱图峰拟合曲线Fig.2 The fitting curves for FT-IR spectra of the peak
A峰2——分峰范围内峰2曲线底部包围面积,峰位一般在1720 cm-1左右,为氢键化峰
A峰3——分峰范围内峰3曲线底部包围面积,峰位一般在1700 cm-1左右,为氢键化峰
A峰4——分峰范围内峰4曲线底部包围面积,峰位一般在1660 cm-1左右,为氢键化峰
拟合单峰面积及氢键化程度数据列于表1。从表1可看出,随着硬段含量增加,氢键化程度增大。在聚碳酸酯型聚氨酯中,硬段及软段均存在硬段氢键化加强有利于微相分离,而软段氢键化加强则不利于微相分离。
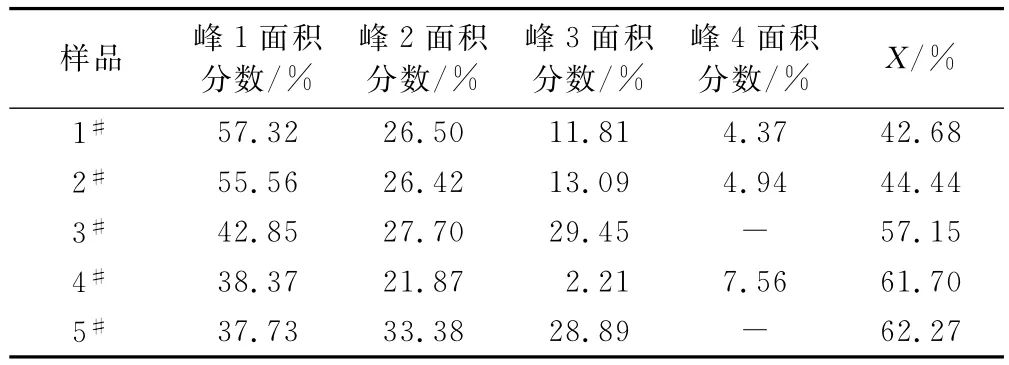
表1 合成的聚氨酯弹性体的C O氢键化程度计算结果Tab.1 Calculation results for C O hydrogen bonds degree of synthetic PU
2.2 DSC分析
DSC测试的第二次升温曲线如图3所示。从图3可以看出,所有样品的Tg均在-37~-20℃之间,均比PCD高,说明合成的聚氨酯弹性体并不是完全相分离,存在一定程度的相混合;其次,合成的聚氨酯弹性体无明显的结晶峰出现,说明所合成的聚氨酯弹性体在室温及体温下均为透明无定形弹性体;第三,随着硬段含量增加,软段的Tg呈逐渐升高的趋势。

图3 原料PCD及合成的聚氨酯弹性体的DSC曲线Fig.3 DSC curves for PCD and synthetic PU elastomers
2.3 DMTA分析
从图4可以看出,随着硬段含量的增加,损耗因子tanδ峰值处温度依次升高,表明软段Tg依次升高,这与DSC测试结果一致;随着硬段含量的增加,tanδ峰值依次降低,而温域逐渐变宽,进一步证实了材料的微相分离程度依次降低。结合红外谱图C O拟合分峰结果,氢键化程度随硬段含量增加而增大,这表明合成的聚氨酯弹性体中软段C O氢键占主导,随硬段含量增加,相容性增加,微相分离程度降低。从图5可以看出,随硬段含量升高,弹性模量依次增大。
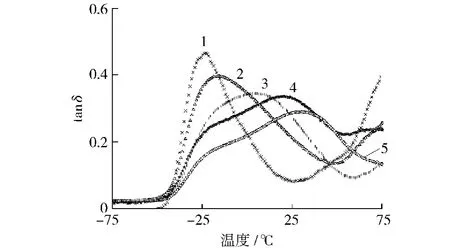
图4 合成的聚氨酯弹性体的损耗因子曲线Fig.4 Loss factor curves for synthetic PU elastomers
2.4 GPC法测相对分子质量
从表2可以看出,合成的聚氨酯弹性体的重均相对分子质量均在35000以上,分散系数在1.82~2.96之间;随硬段含量升高,重均相对分子质量呈先增大后减小趋势,分散系数逐渐增大。这是因为硬段含量较低时,预聚时异氰酸酯过量较少,单体过量分率低,预聚物聚合度高,分散系数小,但预聚物黏度大,扩链时链移动阻力大导致相对分子质量不高;硬段含量较高时,单体过量分率高,预聚物聚合度小,分散系数大,当硬段含量过高时可能存在少量异氰酸酯单体,扩链时扩链剂可与单体异氰酸酯反应,进一步导致相对分子质量降低和分散系数增大。
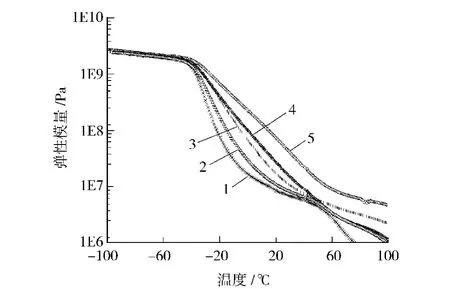
图5 合成的聚氨酯弹性体的弹性模量曲线Fig.5 Elastic modulus curves for synthetic PU elastomers

表2 合成的聚氨酯弹性体的GPC测试结果Tab.2 GPC results for synthetic PU elastomers
2.5 力学性能测试
从图6和表3可以看出,随硬段含量的增加,拉伸强度先增大后减小,断裂伸长率则呈逐渐降低趋势,刚性增强,硬度逐渐升高。拉伸强度变化趋势与相对分子质量变化趋势一致,相对分子质量越大,分子链越长,拉伸时使团状分子整链拉伸并产生相对滑动需要的力越大,因此拉伸强度也越大。
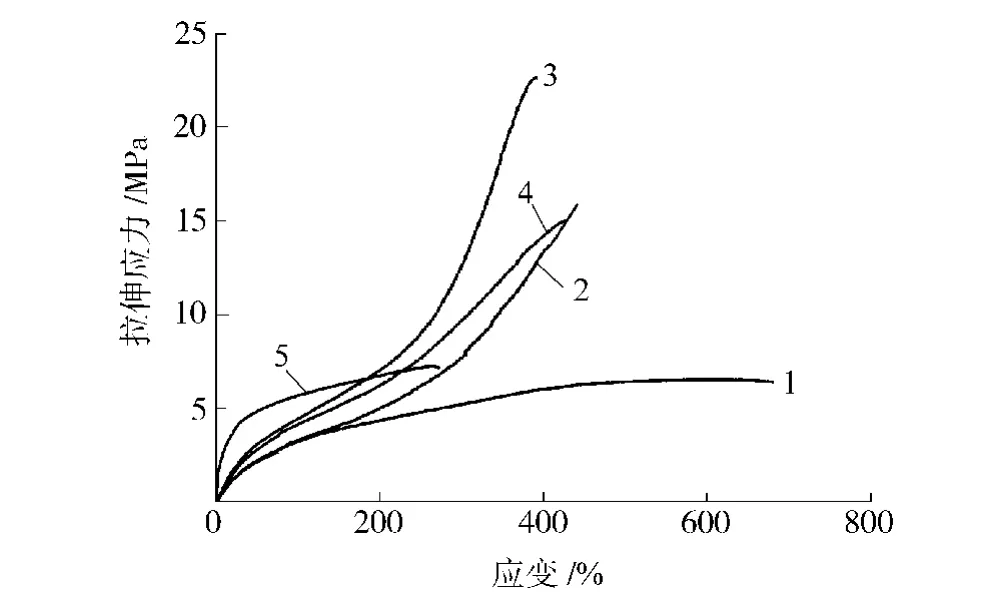
图6 合成的聚氨酯弹性体的拉伸应力应变曲线Fig.6 Tensile stress-strain curves for synthetic PU elastomers
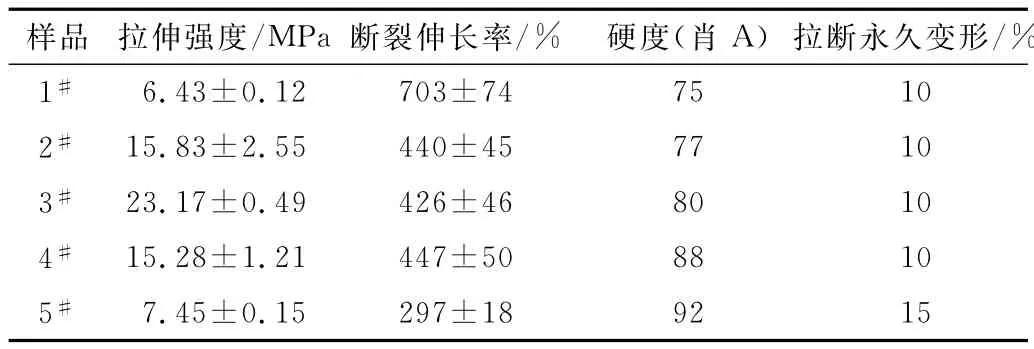
表3 合成的聚氨酯弹性体的拉伸性能Tab.3 Tensile properties of synthetic PU elastomers
从图7可以看出,压缩模量随着硬段含量增高而增大,这与拉伸应力应变曲线中模量及硬度变化一致。

图7 合成的聚氨酯弹性体的压缩应力应变曲线Fig.7 Compression stress-strain curves for synthesis PU elastomers
2.6 水解质量损失率、吸水率
从图8和图9可以看出,不同样品浸泡20 d内的质量损失率随时间推移呈逐渐增大趋势,20 d后质量损失率趋于稳定,在0.6%~0.7%左右;浸泡10 d内吸水率随时间推移呈逐渐增大趋势,10 d后吸水率趋于饱和,保持在0.9%~1.1%左右。表明所合成的聚氨酯弹性体存在一定的微降解,但仍具有较好的耐水解性。
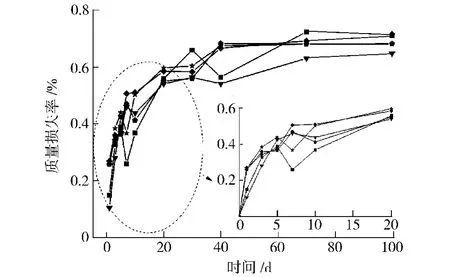
图8 水解不同时间后合成的聚氨酯弹性体的质量损失率Fig.8 Mass loss rate of synthesis PU elastomers with different hydrolysis time

图9 水解不同时间后合成的聚氨酯弹性体的吸水率Fig.9 Water absorption of synthesis PU elastomers with different hydrolysis time
2.7 体外细胞毒性
根据体外细胞毒性试验评级[19],RGR≥100%为0级,75%~99%为1级,50%~74%为2级,25%~49%为3级,0~24%为4级,0为5级。其中0~1级为合格,2级需结合细胞形态综合评定,3~5级为不合格。RGR的计算方式如式(2)所示。

式中RGR——细胞相对增值率,%
OD——光密度,是吸光度的检测单位,取1
从图10可以看出,除3#浸泡144 h毒性为2级外,其他均为0~1级毒性,合格。
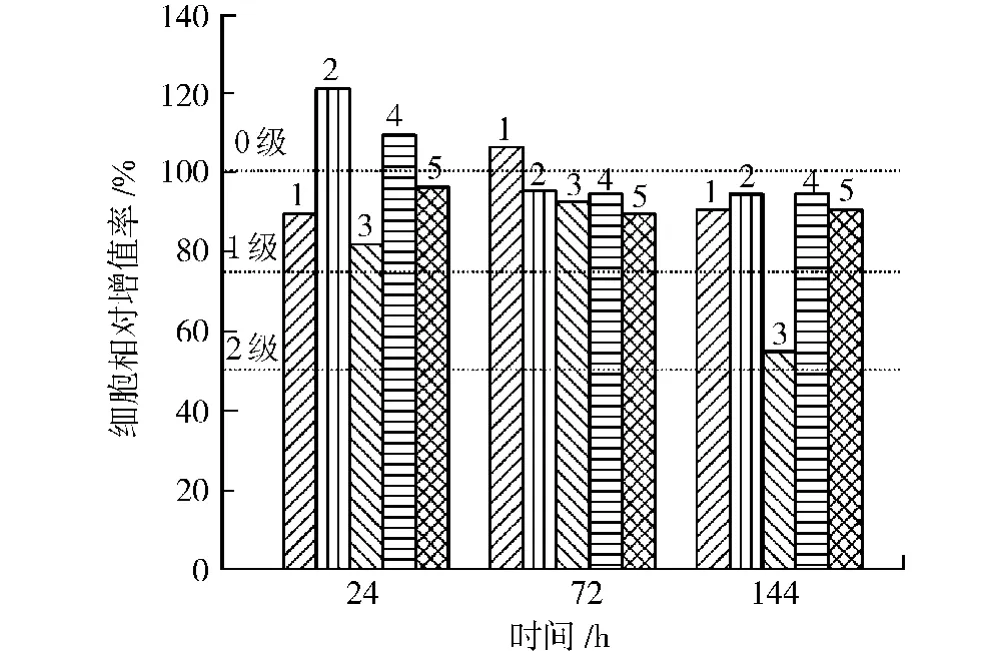
图10 L929细胞相对增值率Fig.10 The relative growth rate of L929 cells
L929细胞在3#样品及阴性对照组材料浸提液中培养144 h后的形态如图11所示。从图11可以看出,相对于对照组来说,3#样品仅细胞密度略减少,细胞形态多成菱形或梭形,形态变化不大,因此为3#样品为低毒材料。
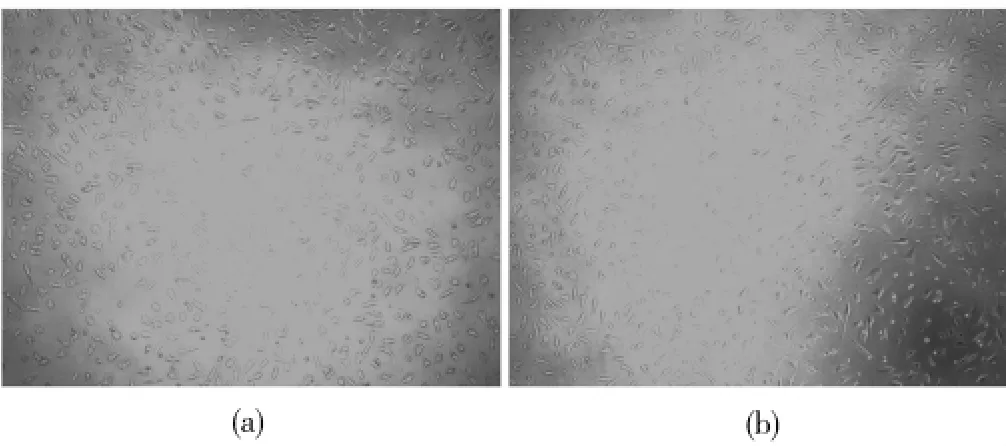
图11 L929细胞培养144 h的细胞形态图(10×)Fig.11 L929 cell morphology after 144 h
3 结论
(1)采用预聚物法合成出了一系列的HMDI基聚碳酸酯型透明的无定形聚氨酯弹性体,系统表征了其微相分离结构及力学性能、水解性能及生物相容性;
(2)随硬段含量增加,合成的聚氨酯弹性体微相分离程度降低;相对分子质量及拉伸强度均呈先增大后减小趋势,分散系数逐渐增大;断裂伸长率呈逐渐降低趋势,刚性、硬度及压缩模量均逐渐增大;
(3)合成的聚氨酯弹性体水解吸水率及质量损失率随时间先增大后趋于饱和,100 d内质量损失率和吸水率均低于1.1%,表明合成的聚氨酯弹性体具有较好的耐水解性;
(4)体外细胞毒性基本为0~1级,表明合成的聚氨酯弹性体具有较好的生物相容性,作为可长期植入体内的生物稳定型医用材料具有良好的应用前景。
[1] Szycher M,Siciliano A A,Reed A M.Polyurethane Elastomers in Medicine[M].NewYork:Marcel Dekker Inc,1994:233-244.
[2] 傅明源,孙酣经.聚氨酯弹性体及其应用[M].北京:化学工业出版社,2006:29-30.
[3] Pinchuk L.A Review of the Biostability and Carcinogenicity of Polyurethanes in Medicine and the NewGeneration of Biostable Polyurethanes[J].Journal of Biomaterials Science:Polymer Edition,1994,6(3):225-267.
[4] LabrowR S,Erfle D J,Santerre J P.Elastase-induced Hydrolysis of Synthetic Solid Substrates:Poly(ester-ureaurethane)and Poly(ether-urea-urethane)[J].Biomaterials,1996,17(24):2381-2388.
[5] Szycher M,Reed A M,Siciliano A A.In Vivo Testing of Biostable Polyurethane[J].Journal of Biomaterials Applications,1991,6(2):110-130.
[6] Zhao Q,TophamN,Anderson J M,et al.Foreign-body Giant Cells and Polyurethane Biostability:In Vivo Correlation of Cell Adhesion and Surface Cracking[J].Journal of Biomedical Materials Research,1991,25(2):177-183.
[7] Tanzi MC,Mantovani D,Petrini P,et al.Chemical Stability of Polyether Urethanes Versus Polycarbonate Urethanes[J].Journal of Biomedical Materials Research,1997,36(4):550-559.
[8] Seifalian A M,Salacinski H J,Tiwari A,et al.In Vivo Biostability of a Poly(carbonate-urea)urethane Graft[J].Biomaterials,2003,24(14):2549-2557.
[9] Zycher M,Reed A M,Siciliano A A.In Vivo Testing of a Biostable Polyurethane[J].Journal of Biomaterials Applications,1991,6(2):110-130.
[10] Szycher M,Reed A M.Medical-grade Polyurethanes:a Critical Review[C].Annual Technical Conference-ANTEC,Conference Proceedings,1996,3:2758-2766.
[11] Tang Y W,LabowR S,Santerre J P.Enzyme-induced Biodegradation of Polycarbonate Polyurethanes:Dependence on Hard-segment Concentration[J].Journal of Biomedical Materials Research Part A,2001,56(4):516-528.
[12] Christenson E M,Dadsetan M,Wiggins M,et al.Poly(carbonate urethane)and Poly(ether urethane)Biodegradation:In Vivo Studies[J].Journal of Biomedical Materials Research Part A,2004,69(3):407-416.
[13] Szycher M,Poirier V L,Dempsey D J.Development of an Aliphatic Biomedical-grade Polyurethane Elastomer[J].Journal of Elastomers and Plastics,1983,15(2):81-95.
[14] Daka J N,Chawla A S.Release of Chemicals from Polyurethane Foamin the Meme Breast Implant[J].Biomaterials Artificial Cells and Immobilization Biotechnology,1993,21(1):23-46.
[15] Tang Y W,LabowR S,Santerre J P.Enzyme-induced Biodegradation of Polycarbonate Polyurethanes:Dependence on Hard-segment Concentration[J].Journal of Biomedical Materials Research,2001,56(4):516-528.
[16] 薛 燕,冯亚凯,刘晓建.聚碳酸酯型聚氨酯材料的制备和性能研究[J].聚氨酯工业,2007,22(1):21-23.Xue Yan,Feng Yakai,Liu Xiaojian.Research on Preparation and Properties of Polycarbonate Polyurethane Material[J].Polyurethane Industry,2007,22(1):21-23.
[17] 刘益军.聚氨酯原料及助剂手册[M].北京:化学工业出版社,2005:21-23.
[18] 宁超峰,王得宁,胡春圃.聚氨酯弹性体的形态结构和电性能研究不同起始反应温度的影响[J].功能高分子学报,2000,13(3):261-263.Ning Cha ofeng,Wang Dening,Hu Chunpu.Morphology and Electrical Insulating Property of Polyurethane Elastomers Infulunce of Different Starting Reaction Temperatures[J].Journal of Functional Polymers,2000,13(3):261-263.
[19] 中国标准化技术委员会.GB/T 16886.5—2003医疗器械生物力学评价.第5部分:体外细胞毒性试验[S].北京:中国标准出版社,2003.
