梓木内酯的优化合成和细胞毒活性研究*
2012-01-08赵建春李英霞
赵建春,丁 宁,张 伟,王 鹏,李英霞**
(1.中国海洋大学医药学院海洋药物教育部重点实验室,山东青岛266003;2.复旦大学药学院,上海201203)
梓木内酯的优化合成和细胞毒活性研究*
赵建春1,丁 宁2,张 伟2,王 鹏1,李英霞2**
(1.中国海洋大学医药学院海洋药物教育部重点实验室,山东青岛266003;2.复旦大学药学院,上海201203)
以2-甲酰基苯甲酸为原料,通过Wittig反应、内酯化、反麦克尔加成、一锅法苯硒化-氧化消除引入双键4步反应,以18%的总收率合成梓木内酯。采用1HNMR、13CNMR、MS、1H-1H COSY、NOE等波谱数据确证了中间体的结构及11,12a,12b的立体构型。与已知方法相比,此合成方法具有原料易得、反应条件温和、操作简单、收率良好等特点。采用SRB染色法对其进行2种肿瘤细胞株(MCF-7,BxPC3)的细胞毒活性测试,发现梓木内酯具有中等强度的抗肿瘤活性。
梓木内酯;一锅法;立体构型;合成;细胞毒活性
梓树(又名黄金树),属紫葳科,广泛分布于日本和中国的南部地区。在民间,梓树的果实用来治疗慢性间质性肾炎、蛋白尿、水肿,根和树皮用来治疗发烧、黄疸、肾炎引起的水肿,叶子用来治疗烫伤[1-3]。1965年,Nagakura及其同事首次从梓树的心材中提取分离到梓木内酯[1]。最近研究结果表明,梓木内酯具有抗病毒的活性[4]、能增加3,4-L-二羟基苯丙氨酸诱导的细胞毒活性[5]。另外,梓木内酯还能抑制多巴胺的合成[5]、抑制脂多糖诱导巨噬细胞中NO的生成[6]。
天然存在的梓木内酯为外消旋体[7]。目前已有3个研究组报道了其合成方法,化合物硒化物4是共同的关键中间体(Scheme 1)。Marx[8]等首先报道了梓木内酯的全合成,在二异丙基氨基锂(LDA)作用下2-苯硒基-5,5-二甲基戊内酯(2)与3-溴苯酞(3)偶联制备硒化物4,再氧化消除苯硒基得到梓木内酯,偶联收率仅为47%。Pinder[7]将苯酞的3-Br换成更易于离去的3-OTs,与5,5-二甲基戊内酯(6)进行偶联,以78%的收率得到氢化梓木内酯7,但由氢化梓木内酯7引入苯硒基制备硒化物4的收率仅有15%。Narasimhan[9]以2-甲酰基苯甲酸(8)为原料,与磷叶立德9进行Wittig反应得到内酯10,在TMSCl/NaI作用下环化得到内酯7,然后选择性打开五元内酯环并在不饱和内酯11上引入苯硒基得硒化物4,最后采用H2O2氧化消除苯硒基得到梓木内酯。3种合成路线中,Narasimhan路线收率较高,但是步骤较长,操作较麻烦。
综合比较以上3条合成路线的优缺点,本文在Narasimhan路线的基础上,优化了梓木内酯的全合成,最终以4步反应、18%的总收率得到了梓木内酯(Scheme 2),是目前报道的最有效的一种方法,并且通过NOE确定了关键中间体的立体构型。
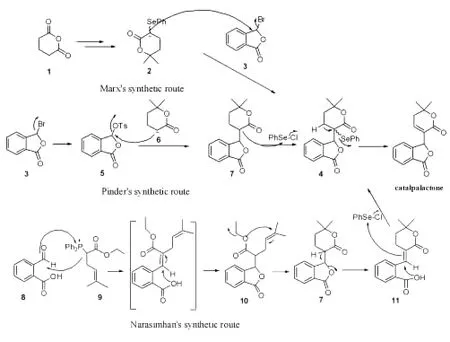
图1 合成梓木内酯的3种路线Fig.1 Three synthesis routes of catalpalactone
本文将梓木内酯进行了体外抗肿瘤测试,肿瘤细胞株选用人乳腺癌细胞(MCF-7),人胰腺癌细胞(Bx-PC3),显示了中等程度的抗肿瘤活性(Table 1)。
1 实验部分
1.1 试剂和仪器
上海精密科学仪器有限公司X-4显微熔点仪,温度计未校正;Varian/Mercury Plus 400MHz核磁共振波谱仪,四甲基硅烷(TMS)为内标;Agilent 1100LC/MSD型质谱仪;JASCO P-1020自动比旋光仪;PhSeCl为Acros公司产品,其余均为国产AR级试剂,其中CH2Cl2、THF经CaH2回流重蒸处理,CH3CN经五氧化二磷重蒸处理。柱层析硅胶(200~300目)由青岛海洋化工厂生产;薄层用预制硅胶板(TLC,GF254)烟台江友硅胶开发有限公司生产。
1.2 化合物10,12a,12b的合成方法
磷叶立德9[9](9.16g,22mmol)溶在150mL氯仿中,加入2-甲酰基苯甲酸(3.3g,22mmol),加热回流4h。TLC跟踪反应完毕后,减压蒸除溶剂,加入乙酸乙酯(100mL)稀释,饱和食盐水(50mL)洗涤,无水Na2SO4干燥,减压浓缩得到黏稠液体,柱层析(EtOAc∶Petroleum ether,1∶20)依次得到化合物10,12a,12b。回收2-甲酰基苯甲酸0.956g。
Ethyl 5-methyl-2-(3-oxo-1,3-dihydroisobenzofuran-1-yl)hex-4-enoate(10)
无色黏稠透明液体512.4mg,产率8%;[α]20D-68.5(c 0.20,CHCl3);1HNMR(DMSO-d6,400 MHz)δ:7.56-7.84(m,8H,Ar),5.88(d,1H,J=5.2Hz,OCHAr),5.77(d,1H,J=4.0Hz,OC HAr),5.11(t,1H,J=7.4Hz,C),4.93(t,1H,J=7.3Hz,CH),4.09(m,2H,OCH2CH3),3.78(m,2H,OCH2CH3),3.18-3.08(m,2H,COCHCH2),2.47(m,2H,COCHCH2),2.30(m,1H,COCHCH2),2.08(m,1H,COCHCH2),1.63(s,3H,CCH3),1.54(s,3H,CCH3),1.51(s,3H,CCH3),1.36(s,3H,CCH3),1.14(t,3H,J=7.0Hz,CH2CH3),0.79(t,3H,J=7.0Hz,CH2CH3);ESI-MS m/z:289.1([M+H]+)。
(Z)-2-(2-(Ethoxycarbonyl)-5-methylhexa-1,4-dienyl)benzoic acid(12a)
黄色黏稠油状物1.596g,产率25%;1HNMR(DMSO-d6,400MHz)δ:7.87(dd,1H,J=1.2,7.8 Hz,ArH),7.46(td,1H,J=1.2,7.4Hz,ArH),7.36(t,1H,J=7.4,7.8Hz,ArH),7.19(s,1H,C=CHAr),7.10(d,1H,J=7.8Hz,ArH),5.16(t,1H,J=7.1Hz,CH=C(CH3)2),3.85(m,2H,OCH2-CH3),3.03(d,2H,J=7.1Hz,CH2CH=C(CH3)2),1.68(s,3H,CCH3),1.63(s,3H,CCH3),0.82(t,3H,J=7.2Hz,OCH2CH3);13CNMR(DMSO-d6,100MHz):δ168.0,167.8,138.5,135.3,133.8,131.8,131.6,130.0,129.4,129.1,127.5,120.2,59.7,32.5,25.6,17.6,13.5;ESI-MS m/z:289.1([M+H]+);HRMS calcd for C17H21O4289.1440,found 289.1445。
(E)-2-(2-(Ethoxycarbonyl)-5-methylhexa-1,4-dienyl)benzoic acid(12b)
黄色黏稠油状物1.578g,产率25%;1HNMR(DMSO-d6,400MHz)δ:7.95(s,1H,ArCH=C),7.92(d,1H,J=7.0Hz,ArH),7.60(t,1H,J=7.4Hz,ArH),7.46(t,1H,J=7.4Hz,ArH),7.26(d,1H,J=7.4Hz,ArH),5.01(t,1H,J=6.6Hz,CH=C(CH3)2),4.18(q,2H,OCH2CH3),2.89(d,2H,J=6.6Hz,CH2CH=C(CH3)2),1.58(s,3H,CCH3),1.37(s,3H,CCH3),1.23(t,3H,J=7.0Hz,OCH2CH3);13CNMR(DMSO-d6,100 MHz):δ167.6,167.2,139.7,136.6,132.0,131.9,130.6,130.4,129.8,128.4,121.5,107.2,60.4,26.4,25.5,17.5,14.2;ESI-MS m/z:289.1([M+H]+);HRMS calcd for C17H21O4289.1440,found 289.1463.
3-(6,6-Dimethyl-2-oxotetrahydro-2H-pyran-3-yl)isobenzofuran-1(3H)-one(7)的合成
化合物10、12a和12b的混合物(312mg,1.08mmol)溶于干燥的CH3CN(15mL)中,搅拌下加入NaI(324 mg,2.16mmol)和TMSCl(276μL,2.16mmol),加热回流18h。减压蒸除溶剂得到褐色油状物,加入水(20 mL),乙醚萃取(20mL×3)。合并有机层,依次用饱和硫代硫酸钠(10mL×2)、饱和食盐水(10mL)洗涤,无水Na2SO4干燥,减压浓缩得到黏稠液体,柱层析(EtOAc∶Petroleum ether,1∶20→1∶10)。依次得到白色固体7A,7B。
化合物7A:白色固体75.5mg,产率27%。m.p.120~121℃;[α]-94.7(c 0.15,CHCl3);1HNMR(DMSO-d6,400MHz)δ:7.82(d,1H,J=7.4Hz,ArH),7.75(td,1H,J=1.0,7.4Hz,ArH),7.58(t,1H,J=7.4,7.8Hz,ArH),7.46(dd,1H,J=0.8,7.8Hz,ArH),6.00(d,1H,J=3.5Hz,OCHAr),3.56-3.51(m,1H,COCHCH2CH2),1.80-1.66(m,3H,CHCH2CH2C(CH3)2),1.30(s,3H,C(CH3)2),1.26-1.28(m,1H,CH2CH2C-(CH3)2),1.11(s,3H,C(CH3)2);ESI-MS m/z:261.1([M+H]+)。
化合物7B:白色固体93.9mg,产率33%。m.p.151~152℃;[α]20D-250.8(c 0.12,CHCl3);1HNMR(DMSO-d6,400MHz)δ:7.84(d,1H,J=7.4Hz,ArH),7.80(t,1H,J=7.4Hz,ArH),7.73(d,1H,J=7.4Hz,ArH),7.58(t,1H,J=7.4Hz,ArH),6.22(d,1H,J=2.3Hz,OCHAr),3.50(m,1H,COCHCH2),1.69-1.64(m,2H,CHCH2-CH2C(CH3)2),1.20(s,3H,C(CH3)2),1.17(s,3H,C(CH3)2),1.16-1.04(m,2H,CHCH2CH2C-(CH3)2);ESI-MS m/z:261.1([M+H]+),回收化合物10(33mg)。
化合物7A:m.p.120~121℃(文献值[9]151~152℃),化合物7B:m.p.151~152℃(文献值[9]118~119℃),熔点测试结果以及谱图数据显示化合物7A与7B极性与文献[9]报道相反。
(E)-2-((6,6-dimethyl-2-oxo-2H-pyran-3(4H,5H,6H)-ylidene)methyl)benzoic acid(11)
化合物7A、7B(20mg,0.077mmol)溶在干燥的THF(2mL)溶液中,加入60%的NaH(3.7mg,0.092 mmol),室温搅拌1h。TLC跟踪反应结束后,蒸除溶剂,加入乙酸乙酯(5mL)稀释,滴加5%的稀醋酸直到水层变成酸性。分离有机层,水层用乙酸乙酯萃取(5 mL×2)。合并有机层,饱和食盐水(3mL)洗涤,无水Na2SO4干燥,减压浓缩得到油状物,柱层析(CH2Cl2∶CH3OH,20∶1)得到化合物(11):白色固体18.6mg,产率93%。m.p.155.0~155.5℃(155℃[9]);1HNMR(CDCl3,400MHz)δ:8.31(s,1H,C=CHAr),8.15(dd,1H,J=1.2,6.7Hz,ArH),7.59(td,1H,J=1.2,6.3Hz,ArH),7.46(t,1H,J=7.4Hz,ArH),7.32(d,1H,J=7.4Hz,ArH),2.60(td,2H,J=2.4,6.8Hz,COCH2CH2-C(CH3)2),1.83(t,2H,J=6.8Hz,CH2CH2C-(CH3)2),1.45(s,6H,C(CH3)2);ESI-MS m/z:261.1([M+H]+)。
3-(6,6-dimethyl-2-oxo-5,6-dihydro-2H-pyran-3-yl)isobenzofuran-1(3H)-one(catalplactone)
化合物11 (40mg,0.154mmol),吡啶(37.1μL,0.461mmol),苯硒基氯(29.4mg,0.154mmol)依次加入到干燥的CH2Cl2(5mL)中,室温搅拌18h。TLC显示反应完毕后,加入2滴冰醋酸,冰浴10min,加入1mL 30%的双氧水,冰浴下继续搅拌1h。TLC检测反应完毕后,加入水,分离有机层,水层用CH2Cl2萃取(5mL×3),合并有机层,饱和食盐水(10mL)洗涤,无水Na2SO4干燥,减压浓缩得到无色半固体,柱层析得到白色固体22.4mg,产率56%。m.p.106~107℃;[α]20D-103.0(c 0.10,CHCl3);1HNMR(CDCl3,400MHz)δ:7.92(d,1H,J=7.6Hz,ArH),7.70-7.53(m,3H,ArH),6.79(t,1H,J=3.3Hz,CHCH2C(CH3)2),6.42(s,1H,COOCHArH),2.58-2.39(m,2H,CHCH2C(CH3)2),1.49(s,3H,CH3),1.35(s,3H,);ESI-MS m/z:259.1([M+H]+)。熔点及1HNMR数据与文献报道相同。
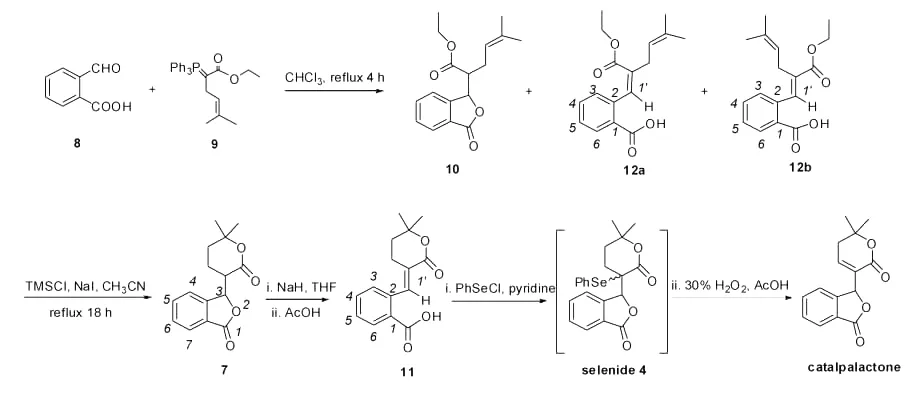
图2 梓木内酯的合成Fig.2 Synthesis of catalpalactone
2 结果与讨论
按照Narasimhan的方法[9],2-甲酰基苯甲酸(8)与磷叶立德9进行Wittig反应后,通过硅胶柱层析分离得到了中性产物10(8%)、酸性产物12a(25%)和12b(25%)3种产物,从产率上计算12a∶12b为1∶1。而文献报道的产物是化合物10(65%)与12a和12b的混合物(20%),并且未将12a和12b分离和结构确证。通过多次实验,严格按照文献报道的反应条件以及控制反应时间都未能重复出文献的结果,均得到主产物为五元内酯环未关环的产物12,并且从反应机理上推断12为10的关环前体。将12a和12b进行了硅胶柱层析分离和结构确证。1HNMR数据显示,极性较小的化合物12a中OCH2CH3质子信号在δ3.85,0.82处,而化合物12b中OCH2CH3质子信号分别在δ4.18,1.23处,推断可能由于苯环的屏蔽效应使化合物12a中OCH2CH3质子信号移向高场区。进一步NOE实验,照射化合物12a中δ3.03处的烯丙位亚甲基,在δ5.16(CH=C(CH3)2),7.19(C=CHAr),1.63(CCH3)处有增益,确证化合物12a为Z-构型。通过NOE实验,照射化合物12b中δ2.89处的烯丙位亚甲基,在δ7.26(3-ArH),5.01(CH=C(CH3)2),1.37(CCH3)处有增益,确证化合物12b为E-构型。这也解释了化合物12b中,异戊烯上的一个甲基由于苯环的屏蔽作用移向高场区δ1.37处(另一个甲基在δ1.58处),1′-H信号由于酯羰基的去屏蔽效应移向低场区δ 7.95(化合物12a中,1′-H信号在δ7.87处),并且E-构型的极性大于Z-构型。
Narasimhan小组用化合物10合成7。推测未关环产物12a和12b在TMSCl/NaI作用下进行内酯化也能得到双关环目标产物。实验结果表明分别用10,12a,12b在TMSCl/NaI作用下进行内酯化,每个化合物都得到双关环产物7(2组非对映异构体混合物7A和7B)。这样Wittig反应后,10、12a、12b不需分离直接进行下一步内酯化反应得到化合物7。这一发现减少了分离操作,用“副产物”同样可以得到期望的双关环产物。
化合物7与NaH在THF溶液中反应,用冰醋酸酸化后以93%的高产率得到唯一的化合物11,在化合物11的1HNMR中,1′-H在δ8.31处,移向低场区,可能是由于六元内酯环的羰基的去屏蔽作用的影响,推测为E-构型。通过NOE实验,照射δ2.60处的烯丙位亚甲基,在δ7.32(3-ArH),1.83(CH2CH2C-(CH3)2),1.45(C-CH3)处有增益,说明酯羰基与1′-H处于同侧,照射δ8.31处的双键氢没有任何增益,从而确证化合物11为E-构型。这也说明这步反应是热力学控制的,得到热力学稳定的E-构型产物。
苯硒基氯在吡啶催化下与化合物(E)-11加成,以42%的收率得到硒化产物4,并同时生成了23%的目标化合物梓木内酯。由于苯硒基极容易被氧化,室温搅拌18h过程中部分苯硒基与空气中氧结合而氧化脱除生成双键。该实验结果提示,(E)-11引入苯硒基后可采用一锅法直接氧化消除苯硒基,而无需分离中间体硒化物4(两组非对映异构体)。因此,在实验过程中,在制得苯硒基化产物4后,在反应液中直接滴加醋酸和双氧水,结果以56%的总收率制得梓木内酯(文献两步收率45%)。本合成条件简化了实验操作,提高了收率。
本文将梓木内酯进行了体外抗肿瘤测试,选用的评估方法为SRB染色法,肿瘤细胞株选用人乳腺癌细胞(MCF-7),人胰腺癌细胞(BxPC3),化合物显示了中等程度的抗肿瘤活性(Table 1)。

表1 梓木内酯对人肿瘤细胞株(MCF-7,BxPC3)的半数生长抑制浓度(IC50)Table 1 Inhibitory effect of catalpalactone response for 50%growth inhibition(IC50)on human tumor cell lines(MCF-7,BxPC3)
3 结语
本文通过对梓木内酯的合成方法进行优化,通过4步反应,以18%的总收率得到梓木内酯。将Wittig反应生成的3种产物10、12a、12b无需分离直接进行下一步的内酯化反应得到化合物7。采用一锅法苯硒化-氧化消除形成梓木内酯的双键,避免了分离纯化的步骤,两步总收率从45%提高到56%。与已知方法相比,此合成方法具有原料易得,操作简单,反应条件温和,收率良好等特点。本研究运用1HNMR、1H-1H COSY、NOE等波谱数据首次确证了含双键的中间体11,12a,12b的立体构型。除此之外,本文首次发现梓木内酯具有抗肿瘤的活性。
[1] Inouye H,Okuda T,Hirata Y,et al.The structure of catalpalactone[J].Tetrahedron Lett,1965,18:1261-1264.
[2] Inouye H,Okuda T,Hayaohi T.Quinones and related compounds in higher plants.II.Naphthoquinones and related compounds from Catalpa wood[J].Chem Pharm Bull,1975,23:384-391.
[3] Imamura H,Suda M,Wood extractives.III.Constituents of Catalpa ovata wood[J].Mokuzai Gakkaishi,1962,8:127-130.
[4] Fujiwara A,Mori T,Iida A,et al.Antitumor-Promoting Naphthoquinones from Catalpa ovata[J].J Nat Prod,1998,61:629-632.
[5] Huang H S,Han X H,Wang B Y,et al.Effects of catalpalactone on dopamine biosynthesis and L-DOPA-induced cytotoxicity in PC12cells[J].Environ Toxicol Chem,2008,26:86-91.
[6] Park B M,Hong S S,Lee C,et al.Naphthoquinones from Catalpa ovata and their inhibitory effects on the production of nitric oxide[J].Arch Pharm Res,2010,33:381-385.
[7] Lane K J,Pinder A R.Synthesis of catalpalactone[J].J Org Chem,1982,47:3171-3172.
[8] Marx J N,Dobrowolski P J.Total synthesis of catalpalactone[J].Tetrahedron Lett,1982,23:4457-4460.
[9] Chordia M D,Narasimhan N S.New synthesis of catalpalactone[J].J Chem Soc,Perkin Trans 1,1991,2:371-376.
An Improved Synthesis and Cytotoxicity Activities of Catalpalactone
ZHAO Jian-Chun1,DING Ning2,ZHANG Wei2,WANG Peng1,LI Ying-Xia2
(1.The Key Laboratory of Marine Drugs,Ministry of Education,School of Medicine and Pharmacy,Ocean University of China,Qingdao 266003,China;2.School of Pharmacy,Fudan University,Shanghai 201203,China)
An improved synthesis of catalpalactone was carried out with a total yield of 18%by using 2-formylbenzoic acid as the starting material followed by a four-steps procedure including Wittig reaction,lactonization,retro-Michael addition and a one-pot manipulation to introduce the double bond.The structures of all of the intermediates as well as the stereo-configurations of double bond in compounds 11,12a and 12bwere determined by1HNMR,13CNMR,MS,1H-1H COSY and NOE techniques.This improved synthetic method provides several advantages such as inexpensive and easily available reagents,mild reaction conditions,simple operations,and high yields.Through being evaluated in vitro against a panel of two human tumor cell lines(MCF-7,BxPC3)which were determined by the SRB assays,catalpalactone displayed good activities against MCF-7and BxPC3.
catalpalactone;one-pot synthesis;stereo-configuration;synthesis;cytotoxicity activity
R914.5
A
1672-5174(2012)1-2-127-05
2011-01-24;
2011-11-02
赵建春(1985-),女,硕士生。E-mail:jianchun_017@yahoo.com.cn
**通讯作者:E-mail:liyx417@fudan.edu.cn
责任编辑 徐 环
