AlCl3催化1,1,1-三甲基-2,2,2-三氯二硅烷裂解的理论研究
2012-01-05徐文媛杜瑞焕
徐文媛, 龙 威, 杜瑞焕, 郝 伟, 李 敏
(华东交通大学 化学化工系 江西 南昌 330013)
0 引言
有机硅材料具有耐高温、防潮、绝缘、耐老化等优异性能,现已广泛应用于国民经济的各个领域.我国的有机硅市场自从20世纪50年代开始快速增长,对有机硅材料的要求也越来越高,而目前又缺乏成熟的有机硅工业技术,因此,有机硅材料的工业制法成为科学研究领域的热点[1-4].
目前工业中制取有机硅材料的重要原料是甲基氯硅烷,而在相关的合成路线中,甲基三氯硅烷、三甲基氯硅烷、甲基二氯硅烷等副产物构成大量的高沸物,高沸物的大量积压造成严重安全隐患,浪费资源,并造成污染.高沸物的综合利用是迫在眉睫的科学难题,1,1,1-三甲基-2,2,2-三氯二硅烷是一种稳定的高沸物成分,利用AlCl3催化分解其生成单硅烷是国内外一种通用的方法,但一直稳定性不好[5-6].本文用量子化学中的密度泛函(B3LYP/6-31+G*)结合多种二级微扰理论方法[7-14],对AlCl3催化条件下1,1,1-三甲基-2,2,2-三氯二硅烷的裂解反应进行研究,以补充理论研究的数据,并为进一步实验提供帮助与参考.
1 催化裂解反应通道
有机硅烷的理论计算已有一定的基础[15-16],实验[17]也表明在AlCl3做为催化剂条件下,1,1,1-三甲基-2,2,2-三氯二硅烷的裂解反应可以进行.由于二硅烷中卤素原子的电负性较大,使Si—Cl键具有明显的离子化成分,而Si—Si键容易被其他基团取代发生断裂,生成单硅烷.按照AlCl3进攻1,1,1-三甲基-2,2,2-三氯二硅烷发生的化学键断裂方式不同,裂解反应有2种反应通道如式(1)~(6)所示,其中(1)~(3)为反应通道1,(4)~(6)为反应通道2.
Me3SiSiCl3+AlCl3→AlCl2SiCl3+Me3SiCl,
(1)
AlCl2SiCl3+CHCl3→CHCl2SiCl3+AlCl3,
(2)
CHCl2SiCl3+CHCl3→CHCl2CHCl2+SiCl4,
(3)
Me3SiSiCl3+AlCl3→AlCl2SiMe3+SiCl4,
(4)
AlCl2SiMe3+CHCl3→CHCl2SiMe3+AlCl3,
(5)
CHCl2SiMe3+CHCl3→CHCl2CHCl2+SiClMe3.
(6)
比较得知,反应通道1和2的产物是相同的,2种反应通道同时并存,互相竞争.2种反应通道的中间产物明显不同,其过渡态几何构型与相关的能量数据也是不同的,因此,对2种反应通道中的反应物、中间产物、产物及过渡态进行全优化计算,并在相同的计算水平上进行振动分析,探究何种反应通道更具有优势非常有必要,对实验也具有较大指导意义.
2 计算方法
使用Guassian 03程序中的密度泛函(B3LYP/6-31+G*)方法分别对不同反应通道中的反应物、中间产物及产物的空间构型进行全优化[10].通过振动频率分析势能面上各驻点的性质,确认优化得到的各个集合构型是势能面上的稳定点还是过渡态.选择虚频唯一的驻点来确定反应的过渡态,并用内禀反应坐标(IRC)理论对过渡态进行了跟踪计算,验证了各过渡态与反应物、产物之间的连接关系.为确定更可靠的能量数据,在B3LYP优化构型的基础上,采用多种二级微扰方法计算各驻点的能量,进行了准确的能量校正.通过对各物质的几何构型、能量校正等工作确定了反应过程中化学键的断裂、生成及变化规律,全部的计算工作在华东交通大学计算工作站完成.
3 结果与讨论
3.1 反应通道1机理分析
反应式(1)中催化剂AlCl3直接进攻Si—Si键,避开了三甲基的位阻效应,基团AlCl2与基团SiCl2结合生成产物AlCl2SiCl2.反应式(2)和(3)表明中间产物继续发生裂解,生成1,1,2,2-四氯乙烷.通过全优化计算寻找反应过程的过渡态,发现其力矩常数有且仅有一个负本征值,说明其过渡态是可信的,其3步的过渡态虚振模式中走向产物的振动如图1所示(TS1,TS2,TS3分别对应式(1),(2),(3)中的过渡态).

图1 各步过渡态走向产物的虚振趋势图
TS1中Al原子与Si原子互相靠拢,形成了式(2)的反应物AlCl2SiCl3;TS2中的C原子与Si原子互相靠拢,形成了式(3)的反应物CHCl2SiCl3,并继续发生反应;TS3中的Cl原子与Si原子互相靠拢,形成了产物SiCl4,并完成反应.由MP2/6-31+G*方法优化各反应的反应物、过渡态和产物的能量数据,结合IRC计算结果绘制成内禀坐标曲线如图2所示,图2中数据单位均为kJ·mol-1.
从图2中各步反应的活化能数据可知,第1步正反应的活化能E1(116.288 kJ·mol-1)比其逆反应的活化能Er1(59.045 kJ·mol-1)大,而后两步正反应的活化能E2、E3(206.483 kJ·mol-1、241.656 kJ·mol-1)分别比其逆反应活化能Er2、Er3(431.529 kJ·mol-1、353.039 kJ·mol-1)小的多,证明此反应体系只能向生成物的方向进行,且反应的速度控制步骤为第3步反应.
3步的反应热数据分别为ΔH1(57.243 kJ·mol-1)、ΔH2(-225.045 kJ·mol-1)、ΔH3(-111.383 kJ·mol-1),即表明第1步反应为吸热反应,第2、3步反应为放热反应,且放热反应放出的热量远大于吸热反应所需要吸收的热量,其总反应热为-279.185 kJ·mol-1.
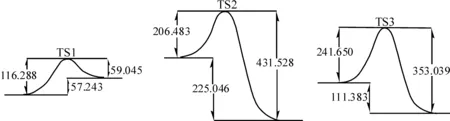
图2 反应通道1中各步反应的内禀曲线图
3.2 反应通道2机理分析
式(4)、(5)、(6)共同组成了反应通道2,其主要为催化剂AlCl3脱去Cl原子,与反应物Me3SiSiCl3中的Si原子结合形成中间产物和终产物SiCl4.将反应式中关键原子编号,并对其过渡态进行了全优化计算,得到如图3的反应过程示意图(其中TS4、TS5、TS6分别为3式的过渡态).

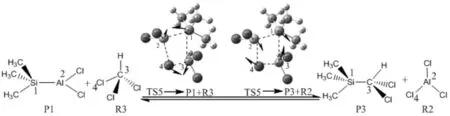

图3 反应过程及过渡态虚振动模式示意图
图3表明3个反应式过渡态都有走向其相应反应物和产物的趋势,进一步证明了过渡态的真实性.由TS4→P1+P2可知,当Si(2)和Al(3)相互靠近的同时,Si(1)和Cl(4)也相互靠近,同时Si(1)和Si(2),Al(3)和Cl(4)相互远离,从而有走向生成物方向的趋势,其他依次类推.根据IRC计算结果得到3个反应中主要原子间距离沿IRC的变化曲线如图4所示.从图4可以看出,从反应物到过渡态再到产物,随着反应的进行,TS4中Si(1)—Cl(4)和Si(2)—Al(3)键、TS5中Al(2)—Cl(4)和Si(1)—C(3)键、TS6中Si(1)—Cl(4)和C(2)—C(3)键逐渐形成,其他主要原子间的距离变化也与过渡态振动模式分析一致.

图4 关键原子间IRC的变化趋势
由MP2/6-31+G*计算所得的反应通道2的内禀反应坐标曲线见图5,图5中数据单位均为kJ·mol-1.由图5可知,反应通道2的第1步正反应的活化能E1(176.927 kJ·mol-1)比其逆反应的活化能Er1(90.964 kJ·mol-1)大,第2步正反应的活化能E2(77.355 kJ·mol-1)比其逆反应活化能Er2(314.951 kJ·mol-1)小很多,第3步正反应的活化能E3(214.913 kJ·mol-1)比其逆反应的活化能Er3(342.481 kJ·mol-1)也小很多,证明了反应只能向正反应方向进行.综合3步反应的活化能可知,此反应体系只能向生成物的方向进行,且反应的速度控制步骤RDS(rate determining step)为式(6)所示的第3步反应.
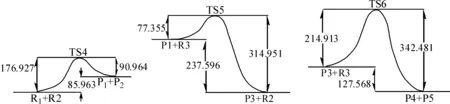
图5 反应通道2中反应内禀曲线图
3步的反应热分别为ΔH4(85.963 kJ·mol-1)、ΔH5(-237.596 kJ·mol-1)、ΔH6(-127.568 kJ·mol-1),表明式(4)所示反应为吸热反应,而式(5)、(6)所示反应为放热反应,且放热反应放出的热量远大于吸热反应所吸收的热量,总反应热为-279.185 kJ·mol-1.采用不同的计算方法对反应通道2的反应进行优化,得知数据结果如表1所示.
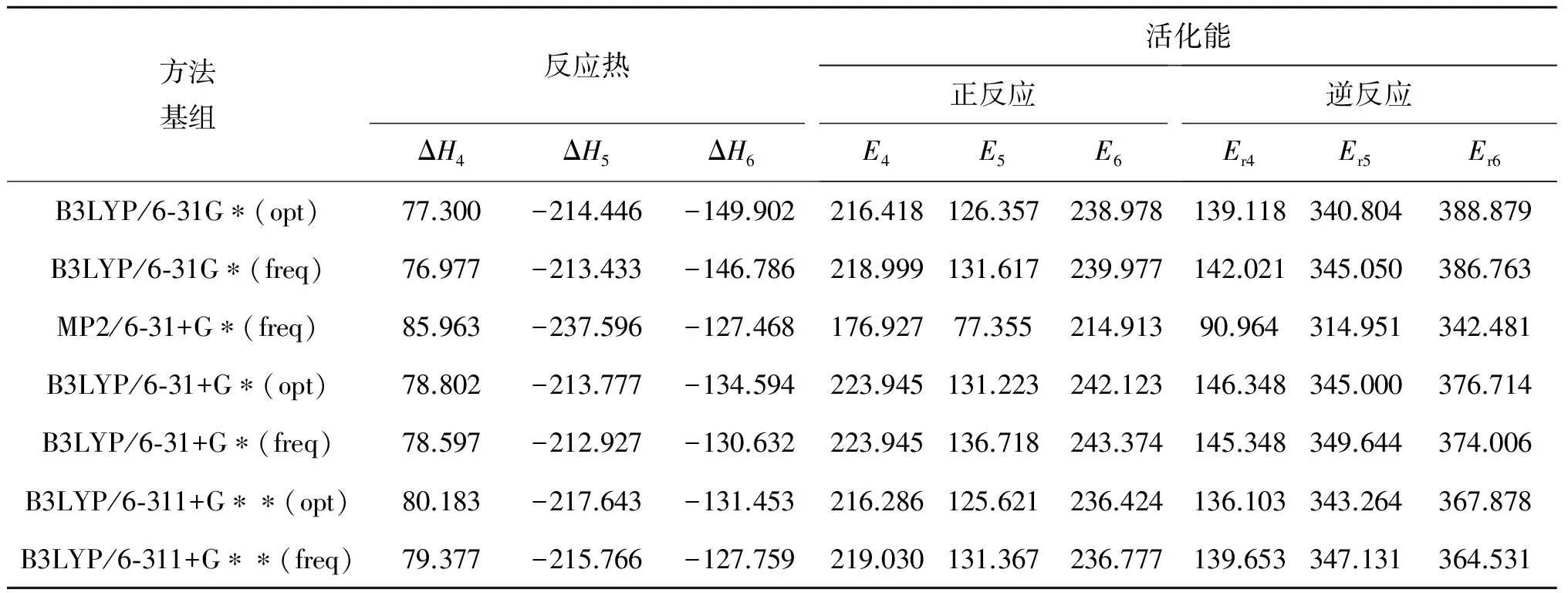
表1 各种方法计算的反应热及活化能
表1中7种方法(基组)计算所得的结果(其中opt表示全优化的结果,freq表示经过振动分析验证后的结果),在能量大小上有差别,但对3个反应的基本特征的描述是一致的,即反应通道2的第1步反应为吸热反应,第2、3步反应为放热反应,反应的速度控制步骤为第3步反应.故可知此反应在常温、常压下,在以AlCl3作为催化剂、CHCl3作为裂解气的条件下较易进行,这与实验结果[17]基本保持一致.
4 结论
1)1,1,1-三甲基-2,2,2-三氯二硅烷在AlCl3催化剂作用下的2种裂解方式可以同时存在,是相互竞争的反应,反应通道2较易进行,总反应为放热反应,其总反应热为-279.185 kJ·mol-1.
2)反应通道1中的3步正反应的活化能分别为116.288 kJ·mol-1,206.483 kJ·mol-1,241.656 kJ·mol-1,反应热分别为57.243 kJ·mol-1,-225.045 kJ·mol-1,-111.383 kJ·mol-1.
3)反应通道2中的3步的正活化能分别为176.927 kJ·mol-1,77.355 kJ·mol-1,214.913 kJ·mol-1,反应热分别为85.963 kJ·mol-1,-237.596 kJ·mol-1,-127.568 kJ·mol-1.

[1] 熊艳锋,张宁.有机高沸物催化裂解制单硅烷催化剂研究进展[J].化工进展,2006,25(8):864-866.
[2] 蔡强,龚红,田健,等.对我国有机硅发展的一些想法[J].现代化工,1997,18(7):3-5.
[3] 章基凯.国内外有机硅材料发展动向与建议[J].化工新型材料,2005,33(11):4-9.
[4] 杨旭石,黄建林,朱凤霞,等.乙基桥联有序介孔有机硅负载Pd(II)有机金属催化剂用于水介质Barbier反应[J].化学学报,2010,68(3):217-222.
[5] Knopf C,Herzog U,Roewer G,et al.Interactions of chloromethyl-disilanes with tetrakis(dimethylamino)ethylene (TDAE),formation of [TDAE]+[Si3Me2Cl7][J].J Organomet Chem,2002,662(1/2):14-22.
[6] Calas R,Dunogues J,Deleris G,et al.Some practical uses of the disilane residue from the direct synthesis of methylchloro-silanes[J].J Organomet Chem,1982,225(1):117-130.
[7] 蔡静,曾薇,李权.8-羟基喹啉过渡金属配合物电子光谱和二阶非线性光学性质的DFT研究[J].化学学报,2009,67(20):2301-2308.
[8] Guirgis G A,Shen Z N,Durig J R,et al.Spectra and structure of silicon containing compounds of the general formula CH2CHSi(CH3)nCl3-n[J].J Mol Struct,1997,410(16):431-434.
[9] Hariharan P C,Pople J A.The effect ofd-functions on molecular orbital energies for hydrocarbons[J].Chem Phys Lett,1974,16(2):217-219.
[10] Pople J A,Gill P M W,Johnson B G.Kohn-Sham density-functional theory within a finite basisset[J].Chem Phys Lett,1992,199(6):557-560.
[11] Simons J,Jorgensen P,Helgaker T U.Higher molecular-deformation derivatives of the configuration-interaction energy[J].Chem Phys Lett,1984,86(3):413-432.
[12] 魏英耐,姚乾凯,徐小树,等.Si(313)表面电子结构特性的理论研究[J].郑州大学学报:自然科学版,2001,33(1): 32-35.
[13] 易春,谢茂浓,彭志坚,等.高温热解聚硅烷制备SiC薄膜初探[J].广西师范大学学报:自然科学版,2000,18(3):20-23.
[14] 熊保库,王林,汤清彬,等.密度泛函方法对BeF分子基态(X2∑+)势能函数的研究[J].郑州大学学报:理学版,2009,41(4):49-52.
[15] 徐文媛,杜瑞焕,龙威,等.AlCl3催化裂解二甲基四氯二硅烷的理论计算[J].浙江大学学报:理学版,2010,37(2): 210-213.
[16] 徐文媛,何忠义,陈玉,等.AlCl3催化歧化制备二甲基二氯硅烷的DFT和MP2研究(II)[J].化学学报,2005,63(16):1474-1478.
[17] 熊艳锋.有机高沸物催化裂解制单硅烷新型催化剂研究及机理探讨[D].南昌:南昌大学,2007.
